Clustal is a series of widely used computer programs used in bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm are also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.
- clustal
- bioinformatics
- algorithm
1. History
There have been many variations of the Clustal software, all of which are listed below:
- Clustal: The original software for multiple sequence alignments, created by Des Higgins in 1988, was based on deriving phylogenetic trees from pairwise sequences of amino acids or nucleotides.[1]
- ClustalV: The second generation of the Clustal software was released in 1992 and was a rewrite of the original Clustal package. It introduced phylogenetic tree reconstruction on the final alignment, the ability to create alignments from existing alignments, and the option to create trees from alignments using a method called Neighbor joining.[2]
- ClustalW: The third generation, released in 1994, greatly improved upon the previous versions. It improved upon the progressive alignment algorithm in various ways, including allowing individual sequences to be weighted down or up according to similarity or divergence respectively in a partial alignment. It also included the ability to run the program in batch mode from the command line.[1]
- ClustalX: This version, released in 1997, was the first to have a graphical user interface.[3]
- ClustalΩ (Omega): The current standard version.[4][5]
- Clustal2: The updated versions of both ClustalW and ClustalX with higher accuracy and efficiency.[6]
The papers describing the clustal software have been very highly cited, with two of them amongst the most cited papers of all time.[7]
The more recent version of the software available for Windows, Mac OS, and Unix/Linux. It is also commonly used via a web interface at its own home page or hosted by the European Bioinformatics Institute.
1.1. Name Origin
The guide tree in the initial programs was constructed via a UPGMA cluster analysis of the pairwise alignments, hence the name CLUSTAL.[8]cf.[9] The first four versions in 1988 had Arabic numerals (1 to 4), whereas with the fifth version Des Higgins switched to Roman numeral V in 1992.[8]cf.[2][10] In 1994 and in 1997, for the next two versions, the letters after the letter V were used and made to correspond to W for Weighted and X for X Window.[8]cf.[3][11] The name omega was chosen to mark a change from the previous ones.[8]
2. Function
All variations of the Clustal software align sequences using a heuristic that progressively builds a multiple sequence alignment from a series of pairwise alignments. This method works by analyzing the sequences as a whole, then utilizing the UPGMA/Neighbor-joining method to generate a distance matrix. A guide tree is then calculated from the scores of the sequences in the matrix, then subsequently used to build the multiple sequence alignment by progressively aligning the sequences in order of similarity.[12] Essentially, Clustal creates multiple sequence alignments through three main steps:
- Do a pairwise alignment using the progressive alignment method
- Create a guide tree (or use a user-defined tree)
- Use the guide tree to carry out a multiple alignment
These steps are carried out automatically when you select "Do Complete Alignment". Other options are "Do Alignment from guide tree and phylogeny" and "Produce guide tree only".
2.1. Input/Output
This program accepts a wide range of input formats, including NBRF/PIR, FASTA, EMBL/Swiss-Prot, Clustal, GCC/MSF, GCG9 RSF, and GDE.
The output format can be one or many of the following: Clustal, NBRF/PIR, GCG/MSF, PHYLIP, GDE, or NEXUS.
| Symbol | Definition | Meaning |
|---|---|---|
| * | asterisk | positions that have a single and fully conserved residue |
| : | colon | conservation between groups of strongly similar properties
with a score greater than .5 on the PAM 250 matrix |
| . | period | conservation between groups of weakly similar properties
with a score less than or equal to .5 on the PAM 250 matrix |
The same symbols are shown for both DNA/RNA alignments and protein alignments, so while * (asterisk) symbols are useful to both, the other consensus symbols should be ignored for DNA/RNA alignments.
2.2. Settings
Many settings can be modified to adapt the alignment algorithm to different circumstances. The main parameters are the gap opening penalty, and the gap extension penalty.
3. Clustal and ClustalV
3.1. Brief Summary
The original program in the Clustal series of software was developed in 1988 as a way to generate multiple sequence alignments on personal computers. ClustalV was released 4 years later and greatly improved upon the original, adding and altering a few key features, including a switch to being written in C instead of Fortran like its predecessor.
3.2. Algorithm
Both versions use the same fast approximate algorithm to calculate the similarity scores between sequences, which in turn produces the pairwise alignments. The algorithm works by calculating the similarity scores as the number of k-tuple matches between two sequences, accounting for a set penalty for gaps. The more similar the sequences, the higher the score, the more divergent, the lower the scores. Once the sequences are scored, a dendrogram is generated through the UPGMA to represent the ordering of the multiple sequence alignment. The higher ordered sets of sequences are aligned first, followed by the rest in descending order. The algorithm allows for very large data sets, and works fast. However, the speed is dependent on the range for the k-tuple matches chosen for the particular sequence type.[13]
3.3. Notable ClustalV Improvements
Some of the most notable additions in ClustalV are profile alignments, and full command line interface options. The ability to use profile alignments allows the user to align two or more previous alignments or sequences to a new alignment and move misaligned sequences (low scored) further down the alignment order. This gives the user the option to gradually and methodically create multiple sequence alignments with more control than the basic option.[12] The option to run from the command line greatly expedites the multiple sequence alignment process. Sequences can be run with a simple command,
clustalv nameoffile.seq
or
clustalv /infile=nameoffile.seq
and the program will determine what type of sequence it is analyzing. When the program is completed, the output of the multiple sequence alignment as well as the dendrogram go to files with .aln and .dnd extensions respectively. The command line interface uses the default parameters, and doesn't allow for other options.[13]
4. ClustalW
4.1. Brief Summary
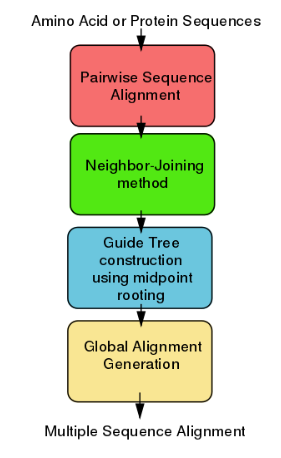
ClustalW like the other Clustal tools is used for aligning multiple nucleotide or protein sequences in an efficient manner. It uses progressive alignment methods, which align the most similar sequences first and work their way down to the least similar sequences until a global alignment is created. ClustalW is a matrix-based algorithm, whereas tools like T-Coffee and Dialign are consistency-based. ClustalW has a fairly efficient algorithm that competes well against other software. This program requires three or more sequences in order to calculate a global alignment, for pairwise sequence alignment (2 sequences) use tools similar to EMBOSS, LALIGN.
4.2. Algorithm
ClustalW uses progressive alignment methods as stated above. In these, the sequences with the best alignment score are aligned first, then progressively more distant groups of sequences are aligned. This heuristic approach is necessary due to the time and memory demand of finding the global optimal solution. The first step to the algorithm is computing a rough distance matrix between each pair of sequences, also known as pairwise sequence alignment. The next step is a neighbor-joining method that uses midpoint rooting to create an overall guide tree.[14] The process it uses to do this is shown in the detailed diagram for the method to the right. The guide tree is then used as a rough template to generate a global alignment.
4.3. Time Complexity
ClustalW has a time complexity of [math]\displaystyle{ O(N^2) }[/math] because of its use of the neighbor-joining method. In the updated version (ClustalW2) there is an option built into the software to use UPGMA which is faster with large input sizes. The command line flag in order to use it instead of neighbor-joining is:
-clustering=UPGMA
For example, on a standard desktop, running UPGMA on 10,000 sequences would produce results in less than a minute while neighbor-joining would take over an hour.[15] By running the ClustalW algorithm with this adjustment, it saves significant amounts of time. ClustalW2 also has an option to use iterative alignment to increase alignment accuracy. While it is not necessarily faster or more efficient complexity-wise, the increase in accuracy is valuable and can be useful for smaller data sizes. These are the various command line flags to achieve this:
-Iteration=Alignment -Iteration=Tree -numiters
The first command line option refines the final alignment. The second option incorporates the scheme into the progressive alignment step of the algorithm. The third specifies the number of iteration cycles where the default value is set to 3.[15]
4.4. Accuracy and Results
The algorithm ClustalW uses provides a close-to-optimal result almost every time. However, it does exceptionally well when the data set contains sequences with varied degrees of divergence. This is because in a data set like this, the guide tree becomes less sensitive to noise. ClustalW was one of the first algorithms to combine pairwise alignment and global alignment in an attempt to be speed efficient, and it worked, but there is a loss in accuracy that other software doesn't have due to this.
ClustalW, when compared to other MSA algorithms, performed as one of the quickest while still maintaining a level of accuracy.[16] There is still much to be improved compared to its consistency-based competitors like T-Coffee. The accuracy for ClustalW when tested against MAFFT, T-Coffee, Clustal Omega, and other MSA implementations had the lowest accuracy for full-length sequences. It had the least RAM memory demanding algorithm out of all the ones tested in the study.[16] While ClustalW recorded the lowest level of accuracy among its competitors, it still maintained what some would deem acceptable. There have been updates and improvements to the algorithm that are present in ClustalW2 that work to increase accuracy while still maintaining its greatly valued speed.[15]
5. Clustal Omega
5.1. Brief Summary
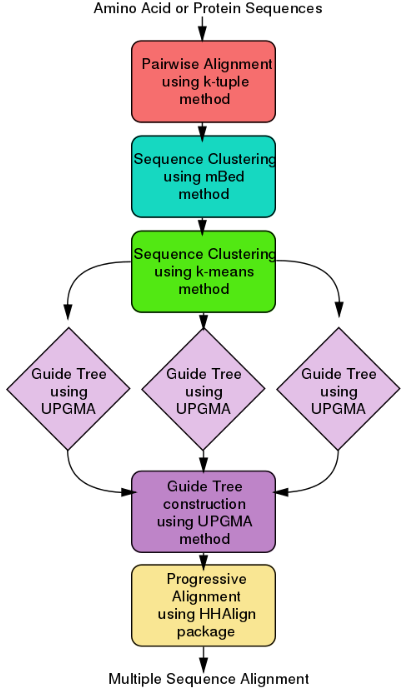
ClustalΩ (alternatively written as Clustal O and Clustal Omega) is a fast and scalable program written in C and C++ used for multiple sequence alignment. It uses seeded guide trees and a new HMM engine that focuses on two profiles to generate these alignments.[17][18] The program requires three or more sequences in order to calculate the multiple sequence alignment, for two sequences use pairwise sequence alignment tools (EMBOSS, LALIGN). Clustal Omega is consistency-based and is widely viewed as one of the fastest online implementations of all multiple sequence alignment tools and still ranks high in accuracy, among both consistency-based and matrix-based algorithms.
5.2. Algorithm
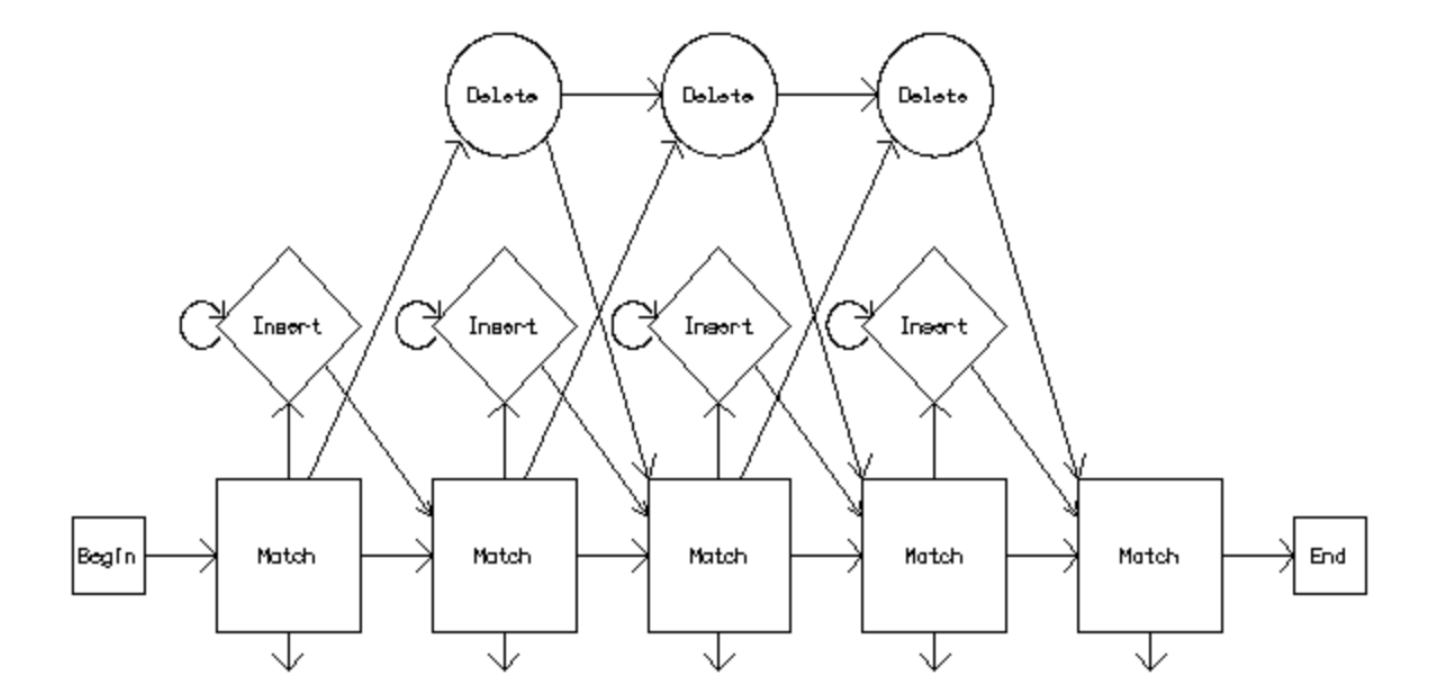
Clustal Omega has five main steps in order to generate the multiple sequence alignment. The first is producing a pairwise alignment using the k-tuple method, also known as the word method. This, in summary, is a heuristic method that isn't guaranteed to find an optimal alignment solution, but is significantly more efficient than the dynamic programming method of alignment. After that, the sequences are clustered using the modified mBed method.[19] The mBed method calculates pairwise distance using sequence embedding. This step is followed by the k-means clustering method. Next, the guide tree is constructed using the UPGMA method. This is shown as multiple guide tree steps leading into one final guide tree construction because of the way the UPGMA algorithm works. At each step, (each diamond in the flowchart) the nearest two clusters are combined and is repeated until the final tree can be assessed. In the final step, the multiple sequence alignment is produced using HHAlign package from the HH-Suite, which uses two profile HMM's. A profile HMM is a linear state machine consisting of a series of nodes, each of which corresponds roughly to a position (column) in the alignment from which it was built.[20]
5.3. Time Complexity
The exact way of computing an optimal alignment between N sequences has a computational complexity of [math]\displaystyle{ O(L^N) }[/math] for N sequences of length L making it prohibitive for even small numbers of sequences. Clustal Omega uses a modified version of mBed which has a complexity of [math]\displaystyle{ O(N \log N) }[/math],[19][21] and produces guide trees that are just as accurate as those from conventional methods. The speed and accuracy of the guide trees in Clustal Omega is attributed to the implementation of a modified mBed algorithm. It also reduces the computational time and memory requirements to complete alignments on large datasets.
5.4. Accuracy and Results
The accuracy of Clustal Omega on a small number of sequences is, on average, very similar to what are considered high quality sequence aligners. The difference comes when using large sets of data with hundreds of thousands of sequences. In these cases, Clustal Omega outperforms other algorithms across the board. Its completion time and overall quality is consistently better than other programs.[22] It is capable of running 100,000+ sequences on one processor in a few hours.
Clustal Omega uses the HHAlign package of the HH-Suite, which aligns two profile Hidden Markov Models instead of a profile-profile comparison. This improves the quality of the sensitivity and alignment significantly.[22] This, combined with the mBed method, gives Clustal Omega its advantage over other sequence aligners. The results end up being very accurate and very quick which is the optimal situation.
On data sets with nonconserved terminal bases, Clustal Omega may be more accurate than Probcons and T-Coffee despite the fact that both of these are consistency-based algorithms, in contrast to Clustal Omega. On an efficiency test with programs that produce high accuracy scores, MAFFT was the fastest, closely followed by Clustal Omega. Both were faster than T-Coffee, however, MAFFT and Clustal Omega required more memory to run.[16]
6. Clustal2 (ClustalW/ClustalX)
Clustal2 is the packaged release of both the command-line ClustalW and graphical Clustal X. Neither are new tools, but are updated and improved versions of the previous implementations seen above. Both downloads come precompiled for many operating systems like Linux, Mac OS X and Windows (both XP and Vista). This release was designed in order to make the website more organized and user friendly, as well as updating the source codes to their most recent versions. Clustal2 is version 2 of both ClustalW and ClustalX, which is where it gets its name. Past versions can still be found on the website, however, every precompilation is now up to date.
The content is sourced from: https://handwiki.org/wiki/Software:Clustal
References
- "CLUSTAL: a package for performing multiple sequence alignment on a microcomputer". Gene 73 (1): 237–44. December 1988. doi:10.1016/0378-1119(88)90330-7. PMID 3243435. https://dx.doi.org/10.1016%2F0378-1119%2888%2990330-7
- "CLUSTAL V: improved software for multiple sequence alignment". Computer Applications in the Biosciences 8 (2): 189–91. April 1992. doi:10.1093/bioinformatics/8.2.189. PMID 1591615. https://dx.doi.org/10.1093%2Fbioinformatics%2F8.2.189
- "The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools". Nucleic Acids Research 25 (24): 4876–82. December 1997. doi:10.1093/nar/25.24.4876. PMID 9396791. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=147148
- Sievers, Fabian; Higgins, Desmond G. (2014-01-01). Russell, David J. ed (in en). Multiple Sequence Alignment Methods. Methods in Molecular Biology. 1079. Humana Press. pp. 105–116. doi:10.1007/978-1-62703-646-7_6. ISBN 9781627036450. https://dx.doi.org/10.1007%2F978-1-62703-646-7_6
- Sievers, Fabian; Higgins, Desmond G. (2002-01-01) (in en). Clustal Omega. 48. John Wiley & Sons, Inc.. 3.13.1–16. doi:10.1002/0471250953.bi0313s48. ISBN 9780471250951. https://dx.doi.org/10.1002%2F0471250953.bi0313s48
- Dineen, David. "Clustal W and Clustal X Multiple Sequence Alignment". http://www.clustal.org/clustal2/.
- "The top 100 papers". Nature 514 (7524): 550–3. October 2014. doi:10.1038/514550a. PMID 25355343. Bibcode: 2014Natur.514..550V. https://dx.doi.org/10.1038%2F514550a
- Des Higgins, presentation at the SMBE 2012 conference in Dublin.
- "CLUSTAL: a package for performing multiple sequence alignment on a microcomputer". Gene 73 (1): 237–44. December 1988. doi:10.1016/0378-1119(88)90330-7. PMID 3243435. https://dx.doi.org/10.1016%2F0378-1119%2888%2990330-7
- "Fast and sensitive multiple sequence alignments on a microcomputer". Computer Applications in the Biosciences 5 (2): 151–3. April 1989. doi:10.1093/bioinformatics/5.2.151. PMID 2720464. https://dx.doi.org/10.1093%2Fbioinformatics%2F5.2.151
- "CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice". Nucleic Acids Research 22 (22): 4673–80. November 1994. doi:10.1093/nar/22.22.4673. PMID 7984417. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=308517
- "CLUSTAL W Algorithm". https://www-bimas.cit.nih.gov/clustalw/clustalw.html.
- https://www.aua.gr/~eliop/mathimata/molevol/Askhsh1/clustalv.htm
- "About CLUSTALW". https://www.megasoftware.net/web_help_7/hc_clustalw.htm.
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M. et al. (2007-09-10). "Clustal W and Clustal X version 2.0" (in en). Bioinformatics 23 (21): 2947–2948. doi:10.1093/bioinformatics/btm404. ISSN 1367-4803. PMID 17846036. https://dx.doi.org/10.1093%2Fbioinformatics%2Fbtm404
- "Assessing the efficiency of multiple sequence alignment programs". Algorithms for Molecular Biology 9 (1): 4. March 2014. doi:10.1186/1748-7188-9-4. PMID 24602402. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4015676
- EMBL-EBI. "Clustal Omega < Multiple Sequence Alignment < EMBL-EBI" (in en). https://www.ebi.ac.uk/Tools/msa/clustalo/.
- Dineen, David. "Clustal Omega, ClustalW and ClustalX Multiple Sequence Alignment". http://www.clustal.org/.
- "Sequence embedding for fast construction of guide trees for multiple sequence alignment". Algorithms for Molecular Biology 5: 21. May 2010. doi:10.1186/1748-7188-5-21. PMID 20470396. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2893182
- "Profile HMM Analysis". http://www.biology.wustl.edu/gcg/hmmanalysis.html.
- "Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega". Molecular Systems Biology 7 (1): 539. October 2011. doi:10.1038/msb.2011.75. PMID 21988835. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3261699
- Daugelaite, Jurate; O' Driscoll, Aisling; Sleator, Roy D. (2013). "An Overview of Multiple Sequence Alignments and Cloud Computing in Bioinformatics" (in en). ISRN Biomathematics 2013: 1–14. doi:10.1155/2013/615630. ISSN 2090-7702. https://dx.doi.org/10.1155%2F2013%2F615630