Age estimation is a paramount issue in criminal, anthropological, and forensic research. Because of this, several areas of research have focused on the establishment of new approaches for age prediction, including bimolecular and anthropological methods. DNA methylation (DNAm) has arisen as one of the hottest topics in the field. Many studies have developed age-prediction models (APMs) based on evaluation of DNAm levels of many genes in different tissue types and using different methodological approaches. However, several challenges and confounder factors should be considered before using methylation levels for age estimation in forensic contexts.
1. Introduction
Age estimation is a paramount issue in forensic science, being required for the identification process of deceased and living individuals. For deceased ones, including human skeletonized remains, the estimative of age can lead to an exclusion; for living individuals, age estimation is important to solve cases of immigration, cases of minors, or for determination of criminal responsibility, for instance [
1,
2,
3,
4].
Aging, being a complex biological process, is difficult to predict and the use of a multidisciplinary approach can improve the age estimation [
5,
6,
7]. For instance, Shi et al. [
6] demonstrated the power of the combination of the anthropological approach (dental and skeletal ages) with the epigenetic approach using DNA methylation (DNAm) levels. During recent years, several areas of knowledge such as anthropology, odontology, chemistry, genetics, and more recently epigenetics, have been continually focused on the improvement of age estimation research. As a result, new methods in several fields have been proposed. In any case, there is not an elected method or approach for age estimation that can be applied to all the forensic cases (living and deceased individuals), with the same accuracy in all age ranges [
1,
8]. In the past, the evaluation of changes in DNAm levels in age-associated genes has been explored as the epigenetic modification with the strongest potential for age prediction in forensic contexts [
3,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19]. As a result, DNAm levels of many age-correlated genes have been evaluated in different tissue types, proposing the development of many highly accurate age prediction models (APMs) [
20]. Despite the increase in research on epigenetic age or DNAm age estimation (DNAm age) in recent years, this tool for age estimation was not implemented until now in forensic laboratories, essentially because of several challenges and confounding factors that need to be considered in future research.
2. DNA Methylation (DNAm): An Epigenetic Mechanism
Epigenetics is a large area of research nowadays. The main epigenetic features (histone modifications, regulation by non-coding RNAs and DNAm) have been associated with several clinical conditions, such as cancer and Alzheimer’s disease, and forensic issues, including age estimation [
21,
22,
23]. One of the most important bases of aging research is DNAm, which has arisen in recent years as one of the most promising and investigated epigenetic features associated with aging [
3,
12,
15,
17,
18,
19]. DNAm is characterized by the addition of a methyl group (CH
3) to the fifth carbon (5C) position of cytosines in the DNA molecule [
24,
25]. Commonly, the methylation of DNA occurs in dinucleotide CpGs (5′-CpG-3′ cytosine–phosphate–guanine) across the genome. However, there are some CpG sites located at clusters named CpG islands, mainly in the promoter regions of the active genes, in which there is no methylation. During aging, there is a change in the human genome methylation levels: most CpGs across the genome lose methylation, and CpG islands gain methylation [
10,
26,
27,
28,
29,
30]. These changes in the pattern of DNAm can be consistent across the individuals or result from stochastic factors. Based on these consistent alterations in DNAm levels of some genes (age-correlated genes), the first generation of “epigenetic clocks” has arisen in different tissue types [
9,
10,
31,
32,
33].
3. Underling Mechanisms of DNAm Changes with Age
As mentioned previously, with aging several changes in DNAm have been observed: consistent alterations across the individuals (epigenetic clock), due to nonstochastic events [
29,
34]; or non-consistent DNAm changes that lead to a DNAm divergence across individuals due to stochastic or environmental factors, such as smoking habits, alcohol consumption, or physical activity (epigenetic drift) [
16,
29,
35,
36]. Indeed, the level of methylation of the CpG sites across the genome can be influenced by several intrinsic factors, such as sex, age and ancestry, or external factors, such as diet, nutrition, stress, toxin exposure, and lifestyles, being useful to predict many individual epigenomic variations also referred to as the epigenetic fingerprint. These epigenomic marks can be related to many phenotypic aspects such as individual lifestyle, health status, physical appearance and individual age estimation [
37]. Despite these two levels of influence, several studies have been conducted in recent years to investigate the relationship between the chronological age of individuals and DNAm levels of some CpGs located at many genes, such as ELOVL2, FHL2, EDARADD, PDE4C, PENK, CCDC102B, C1orf132, TRIM59, and KLF14 [
16,
19,
20,
38,
39,
40]. Some of these genes showed a positive correlation with age, meaning that DNAm levels increase with the increase in chronological age; others revealed a negative age correlation, in which DNAm patterns decrease with the increase in age (
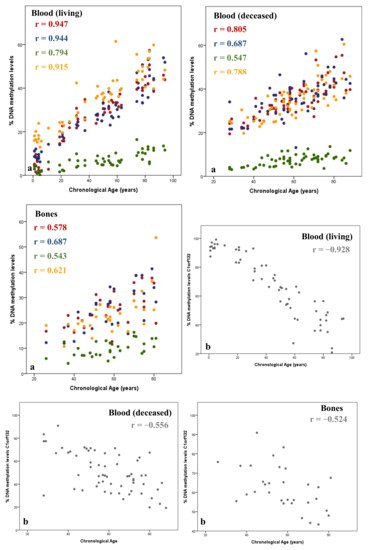
Figure 1).
Figure 1. Positive (
a) and negative (
b) correlations between DNAm levels (%) and chronological age (years) in blood and bone samples. Methylation information is captured through SNaPshot assay in 59 blood samples of living individuals, 62 blood samples of deceased individuals, and 31 bone samples collected during routine autopsies. The corresponding Spearman correlation coefficients (r) are depicted inside each plot. Plots were adapted from ref. [
41].
It should be noted that the same genes or CpGs reveal different patterns of age correlation in different tissues, for instance blood or bone samples, or considering the state of life of the individual in the same tissue type. As shown in
Figure 1, using the same methodology (SNaPshot) for assessing the DNAm levels of the same CpGs from ELOVL2, FHL2, C1orf132, KLF14, and TRIM59, higher age correlation values were obtained for blood samples from living individuals in comparison to the values obtained in bone samples for the same CpGs. This can be related to the tissue specificity of DNAm levels. Several studies have shown different age correlation values for the same genes in different tissues [
9,
42,
43,
44]. This points to the necessity of evaluating the most promising gene in each tissue or building multi-tissue APMs with genes that revealed similar values of correlation among several tissues such as the case of the ELOVL2 gene [
32,
42]. In addition, the state of life of the individual can also influence the correlation with chronological age. In a study developed by our group [
40] it has been shown that age correlation values obtained for ELOVL2, FHL2, C1orf132, KLF14, and TRIM59 genes, captured using the SNaPshot method, were higher in blood samples from living individuals in comparison to age-correlated values captured in blood samples from deceased individuals (
Figure 1). This can be related to postmortem DNAm differences although this issue has not been clarified to date [
40,
45].
4. Methodologies for DNAm Evaluation
Based on the highest age-correlated markers, several authors built many tissue-specific APMs in recent years [
16,
19,
20,
38,
39,
46,
47,
48]. In these studies, the evaluation of DNAm levels is undertaken essentially after sodium bisulfite conversion of genomic DNA. Bisulfite conversion is a chemical modification that allows the easy identification of methylated cytosine and non-methylated cytosine. This treatment with sodium bisulfite leads to the conversion of non-methylated cytosine to uracil, while the methylated cytosine remains as cytosine. Thus, after conversion with sodium bisulfite, followed by amplification using polymerase chain reaction (PCR) and sequencing methods such as SNaPshot, pyrosequencing, Sanger sequencing, or massively parallel sequencing (MPS), the level of methylation in the chromatogram or electropherogram can be quantified [
42,
49,
50].
Of note, it seems that the basics of each methodology can influence DNAm quantification. A recent study by Freire-Aradas and collaborators [
48] showed that depending on the age-correlated gene, the measurement of DNAm levels can be influenced if different methodologies were used. For instance, the DNAm level of the C1orf132 gene is not independent of the methodology used to access to DNAm levels. However, ELOVL2 and FHL2 genes revealed similar patterns of DNAm in EpiTYPER, pyrosequencing, and MiSeq methodologies. Meanwhile, for DNAm levels of ELOVL2, different values were reported between SNaPshot and EpiTYPER or MiSeq methodologies, which can be explained by the use of dyes with different signals intensities in the SNaPshot method, which can require caution in the interpretation of DNAm quantification [
48].
Pyrosequencing has been a widely used method for DNAm evaluation in the past [
5,
10,
43,
51,
52,
53,
54,
55,
56,
57,
58,
59]. Pyrosequencing is easy to use and reveals quantitative data, however, demands high costs [
37]. In recent years, SNaPshot, despite being a semi-quantitative method, has shown promising results in DNAm assessment due to multiplexing analysis [
42,
60,
61,
62]. In addition, the droplet digital PCR (ddPCR) method has also revealed promising results in age estimation allowing to improve age prediction compared to other methodologies [
63]. However, until now only three studies have developed APMs based on this method [
6,
63,
64].
In the near future, it is expected that massively parallel sequencing (MPS), a powerful technology that shows large multiplexing capacities, high sensitivity, and single base resolution, could become a currently used method in forensic contexts for DNAm evaluation. Meanwhile, MPS has been used in a few studies in recent years [
44,
65,
66,
67,
68] showing high model accuracy.
Lastly, considering the methodological aspects of assessment of DNAm levels and the current use of many methodologies for quantification of methylation information, it seems that, in the future, it will be necessary to evaluate the differences between laboratories using the same methodology [
58] and differences between different methodologies [
48] or propose one elected method with standard guidelines to access DNAm levels.
This entry is adapted from the peer-reviewed paper 10.3390/forensicsci2030044