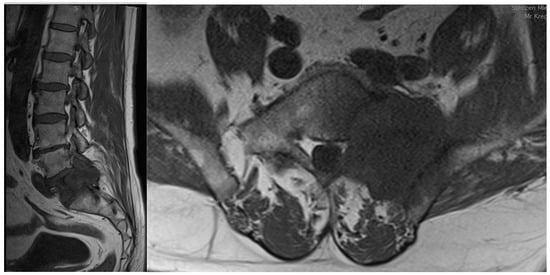
Spine metastases are a common life-threatening complication of advanced-stage malignancies and often result in poor prognosis. Symptomatic spine metastases develop in the course of about 10% of malignant neoplasms. Therefore, it is essential for contemporary medicine to understand metastatic processes in order to find appropriate, targeted therapeutic options. Thanks to continuous research, there appears more and more detailed knowledge about cancer and metastasis, but these transformations are extremely complicated, e.g., due to the complexity of reactions, the variety of places where they occur, or the participation of both tumor cells and host cells in these transitions.
- spine
- metastasis
- tumor
- cancer
1. Introduction
2. Molecular Basis of Spine Metastases
2.1. Escape from the Primary Site
2.2. Cancer Cells Dissemination
2.3. Bone Invasion
2.4. Osteocyte Physiology and Pathology
2.5. Osteoblast Physiology and Pathology
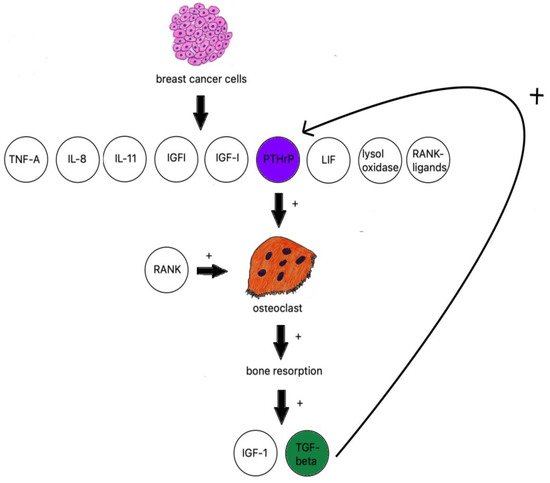
2.6. Osteoblastic Metastasis Pathogenesis
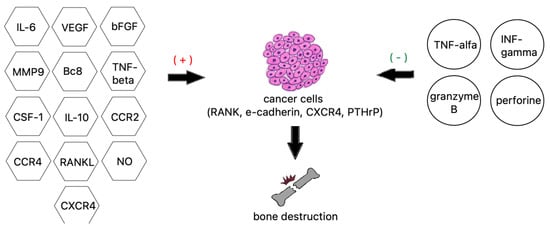
2.7. Osteolytic Bone Metastasis
This entry is adapted from the peer-reviewed paper 10.3390/cancers14194599
References
- Kurisunkal, V.; Gulia, A.; Gupta, S. Principles of Management of Spine Metastasis. Indian J. Orthop. 2020, 54, 181–193.
- Igoumenou, V.G.; Mavrogenis, A.F.; Angelini, A.; Baracco, R.; Benzakour, A.; Benzakour, T.; Bork, M.; Vazifehdan, F.; Nena, U.; Ruggieri, P.; et al. Complications of spine surgery for metastasis. Eur. J. Orthop. Surg. Traumatol. 2020, 30, 37–56.
- Tsukamoto, S.; Kido, A.; Tanaka, Y.; Facchini, G.; Peta, G.; Rossi, G.; Mavrogenis, A.F. Current Overview of Treatment for Metastatic Bone Disease. Curr. Oncol. 2021, 28, 3347–3372.
- Algra, P.R.; Heimans, J.J.; Valk, J.; Nauta, J.J.; Lachniet, M.; van Kooten, B. Do metastases in vertebrae begin in the body or the pedicles? Imaging study in 45 patients. AJR Am. J. Roentgenol. 1992, 158, 1275–1279.
- Ventura, C.; Núñez, S.V.; Gonçalves, A.; Abreu, C.; Costa, L. Bone Health in Metastatic Cancer. Semin. Oncol. Nurs. 2022, 38, 151278.
- Xiao, W.; Zheng, S.; Yang, A.; Zhang, X.; Zou, Y.; Tang, H.; Xie, X. Breast cancer subtypes and the risk of distant metastasis at initial diagnosis: A population-based study. Cancer Manag. Res. 2018, 10, 5329–5338.
- Constans, J.P.; de Divitiis, E.; Donzelli, R.; Spaziante, R.; Meder, J.F.; Haye, C. Spinal metastases with neurological manifestations. Review of 600 cases. J. Neurosurg. 1983, 59, 111–118.
- Coleman, R.E.; Croucher, P.I.; Padhani, A.R.; Clézardin, P.; Chow, E.; Fallon, M.; Guise, T.; Colangeli, S.; Capanna, R.; Costa, L.; et al. Bone metastases. Nat. Rev. Dis. Primers 2020, 6, 83.
- Ziu, E.; Viswanathan, V.K.; Mesfin, F.B. Spinal Metastasis. StatPearls. Published online March 3. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441950/ (accessed on 6 August 2022).
- Maccauro, G.; Spinelli, M.S.; Mauro, S.; Perisano, C.; Graci, C.; Rosa, M.A. Physiopathology of Spine Metastasis. Int. J. Surg.Oncol. 2011, 2011, 107969.
- Papachristou, D.J.; Basdra, E.K.; Papavassiliou, A.G. Bone metastases: Molecular mechanisms and novel therapeutic interventions. Med. Res. Rev. 2012, 32, 611–636.
- Hofbauer, L.C.; Bozec, A.; Rauner, M.; Jakob, F.; Perner, S.; Pantel, K. Novel approaches to target the microenvironment of bone metastasis. Nat. Rev. Clin. Oncol. 2021, 18, 488–505.
- Homayoonfal, M.; Asemi, Z.; Yousefi, B. Potential anticancer properties and mechanisms of thymoquinone in osteosarcoma and bone metastasis. Cell. Mol. Biol. Lett. 2022, 27, 1–28.
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. Int. J. Mol. Sci. 2013, 14, 21087–21113.
- Biologii, P.; Tom, K. Przejście epitelialno-mezenchymalne w procesach nowotworzenia . Postępy Biologii Komórki 2018, 45, 223–236.
- Yin, W.; Wang, J.; Jiang, L.; James Kang, Y. Cancer and stem cells. Exp. Biol. Med. 2021, 246, 1791–1801.
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28.
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027.
- Leong, S.P.; Naxerova, K.; Keller, L.; Pantel, K.; Witte, M. Molecular mechanisms of cancer metastasis via the lymphatic versus the blood vessels. Clin. Exp. Metastasis 2021, 39, 159–179.
- Font-Clos, F.; Zapperi, S.; la Porta, C.A.M. Blood Flow Contributions to Cancer Metastasis. iScience 2020, 23, 101073.
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10.
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed. Res. Int. 2015, 2015, 549412.
- Dudas, J.; Ladanyi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428.
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773.
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412.
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506.
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587.
- Norgard, R.J.; Stanger, B.Z. Isolation and Identification of EMT Subtypes. Methods Mol. Biol. 2021, 2179, 315–326.
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4.
- Liao, T.T.; Yang, M.H. Revisiting epithelial-mesenchymal transition in cancer metastasis: The connection between epithelial plasticity and stemness. Mol. Oncol. 2017, 11, 792–804.
- Li, Q.; Hutchins, A.; Chen, Y.; Li, S.; Shan, Y.; Liao, B.; Zheng, D.; Shi, X.; Li, Y.; Chan, W.Y.; et al. A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat. Commun. 2017, 8, 15166.
- Ban, J.; Fock, V.; Aryee, D.N.T.; Kovar, H. Mechanisms, Diagnosis and Treatment of Bone Metastases. Cells 2021, 10, 2944.
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776.
- Basu, S.; Cheriyamundath, S.; Ben-Ze’ev, A. Cell–cell adhesion: Linking Wnt/β-catenin signaling with partial EMT and stemness traits in tumorigenesis. F1000Research 2018, 7, 1488.
- Zheng, M.; Jiang, Y.-P.; Chen, W.; Li, K.-D.; Liu, X.; Gao, S.-Y.; Feng, H.; Wang, S.-S.; Jiang, J.; Ma, X.-R.; et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget 2015, 6, 6794.
- Li, J.; Riedt, T.; Goossens, S.; García, C.C.; Szczepanski, S.; Brandes, M.; Pieters, T.; Dobrosch, L.; Gütgemann, I.; Farla, N.; et al. The EMT transcription factor Zeb2 controls adult murine hematopoietic differentiation by regulating cytokine signaling. Blood 2017, 129, 460–472.
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118.
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1796.
- Huang, X.; Xiang, L.; Wang, B.; Hu, J.; Liu, C.; Ren, A.; Du, K.; Ye, G.; Liang, Y.; Tang, Y.; et al. CMTM6 promotes migration, invasion, and EMT by interacting with and stabilizing vimentin in hepatocellular carcinoma cells. J. Transl. Med. 2021, 19, 120.
- Huang, H.; Wright, S.; Zhang, J.; Brekken, R.A. Getting a grip on adhesion: Cadherin switching and collagen signaling. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 118472.
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370.
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83.
- Newland, R.; Chan, C.; Chapuis, P.; Keshava, A.; Rickard, M.; Stewart, P.; Suen, M.; Lee, K.; Dent, O. Relative effects of direct spread, lymph node metastasis and venous invasion in relation to blood borne distant metastasis present at the time of resection of colorectal cancer. Pathology 2020, 52, 649–656.
- Amelot, A.; Terrier, L.-M.; Cristini, J.; LeNail, L.-R.; Buffenoir, K.; Pascal-Moussellard, H.; Bonaccorsi, R.; Mathon, B. Approaching spinal metastases spread profile. Surg. Oncol. 2019, 31, 61–66.
- Kumar, N.; Tan, W.L.B.; Wei, W.; Vellayappan, B.A. An overview of the tumors affecting the spine—Inside to out. Neurooncol.Pract. 2020, 7 (Suppl. 1), i10.
- Chen, W.Z.; Shen, J.F.; Zhou, Y.; Chen, X.Y.; Liu, J.M.; Liu, Z.L. Clinical characteristics and risk factors for developing bone metastases in patients with breast cancer. Sci. Rep. 2017, 7, 11325.
- Klusa, D.; Lohaus, F.; Furesi, G.; Rauner, M.; Benešová, M.; Krause, M.; Kurth, I.; Peitzsch, C. Metastatic Spread in Prostate Cancer Patients Influencing Radiotherapy Response. Front. Oncol. 2020, 10, 627379.
- Schmied, L.; Höglund, P.; Meinke, S. Platelet-Mediated Protection of Cancer Cells from Immune Surveillance—Possible Implications for Cancer Immunotherapy. Front. Immunol. 2021, 12, 527.
- Liu, Y.; Zhang, Y.; Ding, Y.; Zhuang, R. Platelet-mediated tumor metastasis mechanism and the role of cell adhesion molecules. Crit. Rev. Oncol. Hematol. 2021, 167, 103502.
- Anvari, S.; Osei, E.; Maftoon, N. Interactions of platelets with circulating tumor cells contribute to cancer metastasis. Sci. Rep. 2021, 11, 15477.
- Akhtar, M.; Haider, A.; Rashid, S.; Al-Nabet, A.D.M.H. Paget’s “Seed and Soil” Theory of Cancer Metastasis: An Idea Whose Time has Come. Adv. Anat. Pathol. 2019, 26, 69–74.
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176.
- Pienta, K.J.; Robertson, B.A.; Coffey, D.S.; Taichman, R.S. The Cancer Diaspora: Metastasis beyond the seed and soil hypothesis. Clin. Cancer Res. 2013, 19, 5849–5855.
- Chernysheva, O.; Markina, I.; Demidov, L.; Kupryshina, N.; Chulkova, S.; Palladina, A.; Antipova, A.; Tupitsyn, N. Bone Marrow Involvement in Melanoma. Potentials for Detection of Disseminated Tumor Cells and Characterization of Their Subsets by Flow Cytometry. Cells 2019, 8, 627.
- Høilund-Carlsen, P.F.; Hess, S.; Werner, T.J.; Alavi, A. Cancer metastasizes to the bone marrow and not to the bone: Time for a paradigm shift! Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 893–897.
- Xiang, L.; Gilkes, D.M. The Contribution of the Immune System in Bone Metastasis Pathogenesis. Int. J. Mol. Sci. 2019, 20, 999.
- Li, H.; Wu, M.; Zhao, X. Role of chemokine systems in cancer and inflammatory diseases. MedComm 2022, 3, e147.
- Fathi, E.; Farahzadi, R.; Valipour, B.; Sanaat, Z. Cytokines secreted from bone marrow derived mesenchymal stem cells promote apoptosis and change cell cycle distribution of K562 cell line as clinical agent in cell transplantation. PLoS ONE 2019, 14, e0215678.
- Chen, P.; Wu, B.; Ji, L.; Zhan, Y. Cytokine Consistency Between Bone Marrow and Peripheral Blood in Patients with Philadelphia-Negative Myeloproliferative Neoplasms. Front. Med. 2021, 8, 598182.
- Florentin, J.; O’Neil, S.P.; Ohayon, L.L.; Uddin, A.; Vasamsetti, S.B.; Arunkumar, A.; Ghosh, S.; Boatz, J.C.; Sui, J.; Kliment, C.R.; et al. VEGF Receptor 1 Promotes Hypoxia-Induced Hematopoietic Progenitor Proliferation and Differentiation. Front. Immunol. 2022, 13, 882484.
- Ganta, V.C.; Choi, M.; Kutateladze, A.; Annex, B.H. VEGF 165 b Modulates Endothelial VEGFR1-STAT3 Signaling Pathway and Angiogenesis in Human and Experimental Peripheral Arterial Disease. Circ. Res. 2017, 120, 282–295.
- Massena, S.; Christoffersson, G.; Vågesjö, E.; Seignez, C.; Gustafsson, K.; Binet, F.; Hidalgo, C.H.; Giraud, A.; Lomei, J.; Weström, S.; et al. Identification and characterization of VEGF-A–responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 2015, 126, 2016–2026.
- Marçola, M.; Rodrigues, C.E. Endothelial progenitor cells in tumor angiogenesis: Another brick in the wall. Stem Cells Int. 2015, 2015, 832649.
- Sanmartin, M.C.; Borzone, F.R.; Giorello, M.B.; Pacienza, N.; Yannarelli, G.; Chasseing, N.A. Bone marrow/bone pre-metastatic niche for breast cancer cells colonization: The role of mesenchymal stromal cells. Crit. Rev. Oncol. Hematol. 2021, 164, 103416.
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317.
- Jelgersma, C.; Vajkoczy, P. How to Target Spinal Metastasis in Experimental Research: An Overview of Currently Used Experimental Mouse Models and Future Prospects. Int. J. Mol. Sci. 2021, 22, 5420.
- Ren, G.; Esposito, M.; Kang, Y. Bone metastasis and the metastatic niche. J. Mol. Med. 2015, 93, 1203.
- Dong, Q.; Liu, X.; Cheng, K.; Sheng, J.; Kong, J.; Liu, T. Pre-metastatic Niche Formation in Different Organs Induced by Tumor Extracellular Vesicles. Front. Cell Dev. Biol. 2021, 9, 2643.
- Marques, C.S.; Santos, A.R.; Gameiro, A.; Correia, J.; Ferreira, F. CXCR4 and its ligand CXCL12 display opposite expression profiles in feline mammary metastatic disease, with the exception of HER2-overexpressing tumors. BMC Cancer 2018, 18, 741.
- Shi, Y.; Riese, D.J.; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 1969.
- Zielińska, K.A.; Katanaev, V.L. The Signaling Duo CXCL12 and CXCR4: Chemokine Fuel for Breast Cancer Tumorigenesis. Cancers 2020, 12, 3071.
- Sun, Y.; Mao, X.; Fan, C.; Liu, C.; Guo, A.; Guan, S.; Jin, Q.; Li, B.; Yao, F.; Jin, F. CXCL12-CXCR4 axis promotes the natural selection of breast cancer cell metastasis. Tumour Biol. 2014, 35, 7765–7773.
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Devel. Ther. 2015, 9, 4953–4964.
- Hamilton, S.L.; Ferando, B.; Eapen, A.S.; Yu, J.C.; Joy, A.R. Cancer Secretome May Influence BSP and DSP Expression in Human Salivary Gland Cells. J. Histochem. Cytochem. 2017, 65, 139–151.
- Zabkiewicz, C.; Resaul, J.; Hargest, R.; Jiang, W.G.; Ye, L. Bone morphogenetic proteins, breast cancer, and bone metastases: Striking the right balance. Endocr. Relat. Cancer 2017, 24, R349–R366.
- Rustamov, V.; Keller, F.; Klicks, J.; Hafner, M.; Rudolf, R. Bone sialoprotein shows enhanced expression in early, high- proliferation stages of three-dimensional spheroid cell cultures of breast cancer cell line MDA-MB-231. Front. Oncol. 2019, 9, 36.
- Chen, F.; Han, Y.; Kang, Y. Bone marrow niches in the regulation of bone metastasis. Br. J. Cancer 2021, 124, 1912–1920.
- Ming, J.; Cronin, S.J.F.; Penninger, J.M. Targeting the RANKL/RANK/OPG Axis for Cancer Therapy. Front. Oncol. 2020, 10, 1283.
- Sisay, M.; Mengistu, G.; Edessa, D. The RANK/RANKL/OPG system in tumorigenesis and metastasis of cancer stem cell: Potential targets for anticancer therapy. OncoTargets Ther. 2017, 10, 3801–3810.
- Litak, J.; Czyzewski, W.; Szymoniuk, M.; Pastuszak, B.; Litak, J.; Litak, G.; Grochowski, C.; Rahnama-Hezavah, M.; Kamieniak, P. Hydroxyapatite Use in Spine Surgery—Molecular and Clinical Aspect. Materials 2022, 15, 2906.
- de Groot, A.F.; Appelman-Dijkstra, N.M.; van der Burg, S.H.; Kroep, J.R. The anti-tumor effect of RANKL inhibition in malignant solid tumors—A systematic review. Cancer Treat. Rev. 2018, 62, 18–28.
- Sobacchi, C.; Menale, C.; Villa, A. The RANKL-RANK Axis: A Bone to Thymus Round Trip. Front. Immunol. 2019, 10, 629.
- Li, B.; Wang, P.; Jiao, J.; Wei, H.; Xu, W.; Zhou, P. Roles of the RANKL–RANK Axis in Immunity—Implications for Pathogenesis and Treatment of Bone Metastasis. Front. Immunol. 2022, 13, 922.
- Kim, K.J.; Lee, Y.; Hwang, H.G.; Sung, S.H.; Lee, M.; Son, Y.J. Betulin Suppresses Osteoclast Formation via Down-Regulating NFATc1. J. Clin. Med. 2018, 7, 154.
- Wang, L.; Chen, K.; He, J.; Kenny, J.; Yuan, Y.; Chen, J.; Liu, Q.; Tan, R.; Zhao, J.; Xu, J. Cytochalasin Z11 inhibits RANKL-induced osteoclastogenesis via suppressing NFATc1 activation. RSC Adv. 2019, 9, 38438–38446.
- Casimiro, S.; Vilhais, G.; Gomes, I.; Costa, L. The Roadmap of RANKL/RANK Pathway in Cancer. Cells 2021, 10, 1978.
- Wang, Y.; Liu, Y.; Huang, Z.; Chen, X.; Zhang, B. The roles of osteoprotegerin in cancer, far beyond a bone player. Cell Death Discov. 2022, 8, 252.
- Marley, K.; Bracha, S.; Seguin, B. Osteoprotegerin activates osteosarcoma cells that co-express RANK and RANKL. Exp. CellRes. 2015, 338, 32–38.
- Tresguerres, F.G.F.; Torres, J.; López-Quiles, J.; Hernández, G.; Vega, J.A.; Tresguerres, I.F. The osteocyte: A multifunctional cell within the bone. Ann. Anat. 2020, 227, 151422.
- Davis, H.M.; Pacheco-Costa, R.; Atkinson, E.G.; Brun, L.R.; Gortazar, A.R.; Harris, J.; Hiasa, M.; Bolarinwa, S.A.; Yoneda, T.; Ivan, M.; et al. Disruption of the Cx43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell 2017, 16, 551–563.
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124.
- Floranović, M.P.; Veličković, L.J. Effect of CXCL12 and Its Receptors on Unpredictable Renal Cell Carcinoma. Clin. Genitourin. Cancer 2020, 18, e337–e342.
- Hiraga, T. Bone metastasis: Interaction between cancer cells and bone microenvironment. J. Oral. Biosci. 2019, 61, 95–98.
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Marini, F.C.; Bussard, K.M. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res. 2019, 21, 31.
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176.
- Suhovskih, A.V.; Kashuba, V.I.; Klein, G.; Grigorieva, E.V. Prostate cancer cells specifically reorganize epithelial cell-fibroblast communication through proteoglycan and junction pathways. Cell Adhes. Migr. 2017, 11, 39.
- Yu, G.; Shen, P.; Lee, Y.-C.; Pan, J.; Song, J.H.; Pan, T.; Lin, S.-C.; Liang, X.; Wang, G.; Panaretakis, T.; et al. Multiple pathways coordinating reprogramming of endothelial cells into osteoblasts by BMP4. iScience. 2021, 24, 102388.
- Ungefroren, H. Autocrine TGF-β in Cancer: Review of the Literature and Caveats in Experimental Analysis. Int. J. Mol. Sci. 2021, 22, 977.
- Wei, X.F.; Chen, Q.L.; Fu, Y.; Zhang, Q.K. Wnt and BMP signaling pathways co-operatively induce the differentiation of multiple myeloma mesenchymal stem cells into osteoblasts by upregulating EMX2. J. Cell. Biochem. 2019, 120, 6515–6527.
- Role of the Wnt/β-Catenin Signaling Pathway in Regulating the Phenotypic Transformation of Aortic Valvular Myofibroblasts to Osteoblast-like Cells. Available online: http://caod.oriprobe.com/articles/48652138/Role_of_the_Wnt_%CE%B2_catenin_signaling_pathway_in_regulating_the_phenotyp.htm (accessed on 6 August 2022).
- Winter, E.M.; Appelman-Dijkstra, N.M. Parathyroid Hormone-Related Protein-Induced Hypercalcemia of Pregnancy Successfully Reversed by a Dopamine Agonist. J. Clin. Endocrinol. Metab. 2017, 102, 4417–4420.
- Goltzman, D. Nonparathyroid Hypercalcemia. Front. Horm. Res. 2019, 51, 77–90.
- Vičić, I.; Belev, B. The pathogenesis of bone metastasis in solid tumors: A review. Croat. Med. J. 2021, 62, 270.
- Kamalakar, A.; Washam, C.L.; Akel, N.S.; Allen, B.J.; Williams, D.K.; Swain, F.L.; Leitzel, K.; Lipton, A.; Gaddy, D.; Suva, L.J. PTHrP(12-48) Modulates the Bone Marrow Microenvironment and Suppresses Human Osteoclast Differentiation and Lifespan. J. Bone Miner Res. 2017, 32, 1421–1431.
- Liang, M.; Ma, Q.; Ding, N.; Luo, F.; Bai, Y.; Kang, F.; Gong, X.; Dong, R.; Dai, J.; Dai, Q.; et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019, 10, 353.
- Reynaud, C.; Ferreras, L.; Di Mauro, P.; Kan, C.; Croset, M.; Bonnelye, E.; Pez, F.; Thomas, C.; Aimond, G.; Karnoub, A.E.; et al. Lysyl oxidase is a strong determinant of tumor cell colonization in bone. CancerRes. 2017, 77, 268–278.
- le Pape, F.; Vargas, G.; Clézardin, P. The role of osteoclasts in breast cancer bone metastasis. J. Bone Oncol. 2016, 5, 93–95.
- Brook, N.; Brook, E.; Dharmarajan, A.; Dass, C.R.; Chan, A. Breast cancer bone metastases: Pathogenesis and therapeutic targets. Int. J. Biochem. Cell Biol. 2018, 96, 63–78.
- Zheng, X.; Kang, W.; Liu, H.; Guo, S. Inhibition effects of total flavonoids from Scutellaria barbata D. Don on human breast carcinoma bone metastasis via down-regulating PTHrP pathway. Int. J. Mol. Med. 2018, 41, 3137–3146.
- Martin, T.J.; Johnson, R.W. Multiple actions of parathyroid hormone-related protein in breast cancer bone metastasis. Br. J.Pharmacol. 2021, 178, 1923–1935.
- Cheng, J.N.; Frye, J.B.; Whitman, S.A.; Kunihiro, A.G.; Pandey, R.; Funk, J.L. A Role for TGFβ Signaling in Preclinical Osteolytic Estrogen Receptor-Positive Breast Cancer Bone Metastases Progression. Int. J. Mol. Sci. 2021, 22, 4463.
- Wu, C.; Chen, M.; Sun, Z.; Ye, Y.; Han, X.; Qin, Y.; Liu, S. Wenshen Zhuanggu formula mitigates breast cancer bone metastasis through the signaling crosstalk among the Jagged1/Notch, TGF-β and IL-6 signaling pathways. J. Ethnopharmacol. 2019, 232, 145–154.
- Meng, X.; Ark, A.V.; Lee, P.; Hostetter, G.; Bhowmick, N.A.; Matrisian, L.M.; Williams, B.O.; Miranti, C.K.; Li, X. Myeloid-specific TGF-β signaling in bone promotes basic-FGF and breast cancer bone metastasis. Oncogene 2016, 35, 2370–2378.
- Das, D.; Hong, J. Prostaglandin E2 Receptor 4 (EP4): A Promising Therapeutic Target for the Treatment of Cancer and Inflammatory Diseases. Curr. Chem. Biol. 2020, 15, 50–68.
- Johnston, K.A.; Lopez, K.M. Lysyl oxidase in cancer inhibition and metastasis. Cancer Lett. 2018, 417, 174–181.
- Vaziri, N.; Shariati, L.; Javanmard, S.H. Leukemia inhibitory factor: A main controller of breast cancer. J. Biosci. 2020, 45, 143.
- Wang, M.; Wang, M.; Wang, Z.; Yu, X.; Song, Y.; Wang, C.; Xu, Y.; Wei, F.; Zhao, Y.; Xu, Y. Long non-coding RNA-CTD-2108O9.1 represses breast cancer metastasis by influencing leukemia inhibitory factor receptor. Cancer Sci. 2018, 109, 1764–1774.
- Hu, G.-F.; Wang, C.; Wu, G.; Zhang, C.; Zhu, W.; Chen, C.; Gu, Y.; Zhang, H.; Yang, Z. AZD3463, an IGF-1R inhibitor, suppresses breast cancer metastasis to bone via modulation of the PI3K-Akt pathway. Ann. Transl. Med. 2020, 8, 336.
- Okamoto, K.; Takayanagi, H. Osteoimmunology. Cold Spring Harb. Perspect. Med. 2019, 9, a031245.