Immunotherapies are promising approaches for treating hepatocellular carcinomas (HCCs) refractory to conventional therapies. However, a recent clinical trial of immune checkpoint inhibitors (ICIs) revealed that anti-tumor responses to ICIs are not satisfactory in HCC cases. Therefore, it is critical to identify molecular markers to predict outcome and develop novel combination therapies that enhance the efficacy of ICIs. Recently, several attempts have been made to classify HCC based on genome, epigenome, and transcriptome analyses. These molecular classifications are characterized by unique clinical and histological features of HCC, as well immune phenotype. For example, HCCs exhibiting gene expression patterns with proliferation signals and stem cell markers are associated with the enrichment of immune infiltrates in tumors, suggesting immune-proficient characteristics for this type of HCC. However, the presence of activating mutations in β-catenin represents a lack of immune infiltrates and refractoriness to ICIs. Although the precise mechanism that links the immunological phenotype with molecular features remains controversial, it is conceivable that alterations of oncogenic cellular signaling in cancer may lead to the expression of immune-regulatory molecules and result in the acquisition of specific immunological microenvironments for each case of HCC. Therefore, these molecular and immune characteristics should be considered for the management of HCC using immunotherapy.
- hepatocellular carcinoma
- molecular classification
- immune phenotype
- immune checkpoint inhibitor
- stem cell marker
- oncogenic signal
- β-catenin
- genetic alteration
1. Introduction
Hepatocellular carcinoma (HCC) remains one of the leading causes of cancer-related morbidity worldwide and generally emerges from a background of chronic liver inflammation [1]. Recent advancements in molecular target therapy have contributed to improvements in the prognosis of HCC patients, even those with advanced disease [2]. However, most cases of HCC show a tolerance or become refractory to molecular target agents during its clinical course [3,4]. On the other hand, immunotherapies are considered to be a promising approach for HCC patients even in those refractory to conventional therapies [5], and several immune components may play a role in the development and progression of this disease [6]. Nevertheless, phase III clinical trials of immune checkpoint monotherapies in patients with HCC have failed to show superiority to control groups for overall survival (OS) and progression-free survival (PFS) [7,8].
Several attempts have been made to subclassify HCC based on genetic and epigenetic alterations [9–12]. It has also been reported that the molecular subclass of HCC sometimes reflects the immune milieu of tumors [13]. For example, an association between molecular alterations of HCC and the expression of immune checkpoint molecules has been reported [14], and alteration of oncogenic signals due to mutations may lead to altered expression of immune modulators [15]. Therefore, a profound understanding of the molecular subclasses that affect the immune status of tumors may provide valuable insight for the rational development of combination therapies using immune checkpoint inhibitors (ICIs).
2. Effective Application of Immune Checkpoint Inhibitors for HCC Cases
2.1. HCC Response to Immune Checkpoint Inhibitors
Although several phase II clinical trials of ICI monotherapies have shown favorable outcomes for the use of ICIs in patients with HCC [48,59], a phase III study failed to demonstrate positive results as the first-line treatment with respect to OS and PFS compared to the multi-kinase inhibitor sorafenib [7], and as the second-line treatment after sorafenib compared to best supportive care (Table 1) [8]. However, there are molecular features that may be associated with response to ICIs. For example, the HCC with microsatellite instability is reported to show good response to treatment with pembrolizumab [60]. The presence of CTNNB1 variants is associated with the activation of Wnt/β-catenin signaling as well as a lack of immune infiltrates in HCC tumors, which are predictors of a poor response to ICIs in patients with HCC [41]. On the other hand, HCC subtypes with high inflammatory infiltrates, such as HCC of the G2 subclass, may be expected for respond to ICIs [34], although additional agents for combination therapy may be required for a good response [40]. Immunohistochemistry-based markers such as CPS may predict the anti-tumor response to ICIs [48]. However, tumor specimens are required in order to perform the immunohistochemical analysis, which are sometimes difficult to obtain in clinical settings. On the other hand, molecular markers based on genetic alterations of tumor cells based on liquid biopsy may be applicable in which DNA from peripheral blood is used for analysis. From this point of view, the development of a mutation-based molecular marker may prove to be a promising approach for identifying responders for ICIs among HCC patients. However, immune infiltrates of tumor tissues frequently express multiple immune checkpoint molecules that are likely to result in refractoriness to immune checkpoint monotherapies [14,34,40]. Therefore, additional agents for combined immune checkpoint blockades should be required to assure improved response rates.
Table 1. Clinical trials and outcomes of immune checkpoint monotherapies in HCC.
|
Clinical Trial ID |
Trial Name |
Agents 1 |
Setting 2 |
Key Outcome 3 |
|
Phase I/II |
|
|
|
|
|
NCT01658878 |
CheckMate 040 |
Nivolumab |
dose-escalation, n = 48, dose-expansion, n = 214 |
ORR: 20% 4 DCR: 64%, (37%) 5 OS: 13.2 months (8.6–NE) 6 |
|
NCT02702414 |
KEYNOTE-224 |
Pembrolizumab |
second-line n = 104 |
ORR: 17% 7 DCR: 62% OS: 12.9 months (9.7–15.5) |
|
Phase III |
|
|
|
|
|
NCT03383458 |
CheckMate 9DX |
Nivolumab versus placebo |
adjuvant, randomized, double-blinded (n = 530) |
RFS |
|
NCT02576509 |
CheckMate 459 |
Nivolumab versus Sorafenib |
first-line, randomized, open label, n = 743 |
Median OS: 16.4 months in the nivolumab group and 14.7 months in the sorafenib group. 8 Median PFS: 3.7 months for nivolumab and 3.8 months for sorafenib. ORR: 15% in the nivolumab group and 7% in the sorafenib group. |
|
NCT03412773 |
Rationale-301 |
Tislelizumab versus sorafenib |
first-line, randomized, open label, (n = 674) |
OS |
|
NCT02702401 |
KEYNOTE-240 |
Pembrolizumab versus placebo |
second-line, randomized, double-blinded, n = 413 |
Median OS: 13.9 months in the pembrolizumab group and 10.6 months in the placebo group; HR 0.781, p = 0.0238. Median PFS: 3.0 months for pembrolizumab and 2.8 months for placebo; HR 0.781, p = 0.0022. 9 ORR: 18.3%, DCR: 62.2% |
1 Bold denotes immune checkpoint inhibitors. 2 n, number of the patients analyzed in the study. The number in the parenthesis shows the number of the planned enrollment. 3 Bold denotes the primary outcome measures of the study. Duration of responses and survival are shown as median values. The numbers in the parenthesis show 95% confidential interval (CI). 4 El-Khoueiy et al. Lancet 2017; 389: 2492–2502 [59]. 5 Disease control with stable disease for ≥6 months. 6 Median overall survival of the sorafenib progressor without viral hepatitis in the dose-expansion cohort. 7 Zhu et al. Lancet Oncol 2018; 19: 940–952 [48]. 8 Yau et al. The European Society for Medical Oncology (ESMO) 2019 congress (# LBA38). 9 Finn et al. J Clin Oncol 2019; 38: 193–202 [8]. The 95% CI of median OS: 11.6 to 16.0 months in the pembrolizumab group and 8.3 to 13.5 months in the placebo group (hazard ratio, HR, 0.781; 95% CI, 0.611 to 0.998; p = 0.0238). The 95% CI of median PFS was 2.8 to 4.1 months for pembrolizumab and 1.6 to 3.0 months for placebo (HR, 0.718; 95% CI, 0.570 to 0.904; p = 0.0022). OS and PFS did not reach statistical significance per specified criteria in this study. ORR, objective response rate; DCR, disease control rate; OS, overall survival; NE, not estimated; RSF, recurrence-free survival; PFS, progression-free survival.
2.2. Combined Immune Checkpoint Blockade Based on Inflammatory Infiltrate Characteristics of HCC
As shown above, several studies have analyzed the expression of immune suppressive receptors and ligands in inflammatory infiltrates [14,34,40,45]. Generally, inflammatory cells in HCC express several immunosuppressive molecules, suggesting that such immune cells are functionally compromised. For example, expression of PD-1, TIM-3, LAG-3, and CTLA4 is significantly higher on CD8+ and CD4+ T-cells in HCC tissue than those in non-tumor tissues or peripheral blood, and dendric cells (DCs), monocytes, and B cells in tumors express ligands for these receptors [45]. In addition, tumor-associated antigen (TAA)-specific CD8+ TILs express higher levels of PD-1, TIM-3, and LAG-3 compared to that of other CD8+ TILs. Importantly, antibodies against PD-L1, TIM-3, or LAG-3 restore responses of HCC-derived T cells to tumor antigens, and treatment with combinations of these antibodies demonstrate additive effects in the restoration of T-cell function response to TAA [45]. On the other hand, Brown et al. reported the resistance of tumor cells to ICIs through the upregulation of IDO in patients with HCC [61]. Both anti-CTLA4 and anti-PD-1 antibodies induce IDO and the combination of ICIs with 1-methyl-D-tryptophan, an inhibitor of IDO, is able to suppress tumor growth of HCC in a mouse model. Therefore, anti-PD-1 therapy combined with anti-TIM-3, anti-LAG-3, or IDO inhibitor may be worth consideration for patients with HCCs that have exhausted immune infiltrates (Figure 1a). In addition to the phase III combined immune checkpoint blockade using anti-PD-1/PD-L1 and anti-CTLA-4 antibodies, currently, phase I/II clinical trials for the combinations of anti-PD-1 and anti-TIM-3 antibodies (ClinicalTrials.gov NCT03680508), anti-PD-1 and anti-LAG-3 antibodies (NCT03250832), and anti-PD-1 antibody and IDO inhibitors (NCT03695250) are ongoing (Table 2).
Table 2. Clinical trials and outcomes of combined immune checkpoint blockade in HCC.
|
Clinical Trial ID |
Trial Name |
Agents 1 |
Setting 2 |
Key Outcome 3 |
|
Phase I/II |
|
|
|
|
|
NCT01658878 |
CheckMate 040 |
Nivolumab + Ipilimumab |
n = 50 |
ORR: 32% 4 DCR: 54% OS: 22.8 months (9.4–NE) DOR: 17.5 months (4.6–30.5) |
|
NCT02519348 |
|
Durvalumab ± Tremelimumab |
n = 40 |
ORR: 25% 5 DCR: 57.5% |
|
NCT03680508 |
|
TSR-002 + TSR-042 (Dostarlimab) |
first-line, (n = 42) |
ORR |
|
NCT03250832 |
|
TSR-033 + TSR-042 |
dose escalation and dose expansion cohorts (n = 200) |
AEs for dose escalation cohort ORR for dose expansion cohort |
|
NCT03695250 |
|
BMS986205 + Nivolumab |
first- or second-line, (n = 23) |
AEs and ORR |
|
Phase III |
|
|
|
|
|
NCT04039607 |
CheckMate9DW |
Nivolumab + Ipilimumab versus Sorafenib/Lenvatinib |
first-line, randomized, open label, (n = 1084) |
OS |
|
NCT03298451 |
HIMARAYA |
Durvalumab ± Tremelimumab versus Sorafenib |
first-line, randomized, open label, (n = 1310) |
OS |
1 Bold denotes immune checkpoint inhibitors. 2 n, number of the patients analyzed in the study. The number in the parenthesis shows the number of the planned enrollment. 3 Bold denotes the primary outcome measures of the study. Duration of responses and survival are shown as median values. The numbers in the parenthesis show 95% confidential interval. 4 Yau et al. J Clin Oncol. 2019; 37 (supplement abstract 4012). 5 Kelley et al. J Clin Oncol 2017; 35 (supplement abstract 4073). DOR, duration of response; AEs, adverse events.
2.3. Combined Blockade of PD-1/PD-L1 and VEGF Axis
Because HCC is known as a hypervascular tumor where the development of tumor vessels plays an important role in its pathogenesis [62,63], several ongoing clinical studies are evaluating the combination of anti-angiogenic agents and ICIs (Table 3) [64]. Multiple agents that target VEGF and its receptor (VEGFR) are proven to be effective in the treatment of HCC, including the anti-VEGFR2 antibody, ramucirumab [65]. In addition, anti-angiogenic agents are believed to alter the immunosuppressive microenvironment in HCC [6]. It has been reported that anti-angiogenesis normalizes the leaky vascular network induced by VEGF, where the lack of adhesion molecules on endothelial cells may impair the extravasation of T cells [62,66] and induce an immune proficient condition. VEGF play a role in the recruitment of Tregs into tumor tissues and M2 polarization of macrophages via the increase of IL-4 and IL-10. VEGF is also critical for inhibition of the maturation of dendric cells (DCs) by activating NF-κB, production of IDO in tumor cell and macrophage, T-cell exhaustion by inducing PD-1, LAG-3 and TIM-3, accumulation of myeloid-derived suppressor cells (MDSCs), and inhibition of natural killer cell activity [6,67]. Therefore, a combination of ICIs with anti-VEGF agents should be effective (Figure 1b) [67–69], although the dosage that best improves the therapeutic effect of ICIs needs to be defined in individual agents [70]. Accordingly, dual blockade of the VEGF/VEGFR and PD-1/PD-L1 axes in patients with advanced HCC using the anti-PD-L1 antibody atezolizumab and the anti-VEGF-A antibody bevacizumab, or the anti-PD-1 antibody camrelizumab and the VRGFR2-TKI apatinib results in considerable ORR (Table 3) [64]. In addition, other combinations modulating immune microenvironment, such as the combination of anti-PD-1 antibody with an inhibitor of TGF-β receptor, is also under the early phase clinical trial (Table 3: NCT02423343).
Table 3. Clinical trials and outcomes of the combination therapies with immune checkpoint inhibitors and molecular targeted agents.
|
Clinical Trial ID |
Trial Name |
Agents 1 |
Setting 2 |
Key Outcome 3 |
|
Phase I/II |
|
|
|
|
|
NCT03299946 |
CaboNivo |
Cabozantinib + Nivolumab |
neoadjuvant, (n = 15) |
AEs and number of patients who complete the treatment. |
|
NCT03006926 |
|
Lenvatinib + Pembrolizumab |
first-line, (dose-escalation, dose-expansion), n = 30 (n = 97) |
ORR: 53.3% (34.3–71.7), DOR: 8.3 months (3.8–11.0) 4 DCR = 90.0%; 73.5–97.9, PFS: 9.7 months 7.7–NE, OS: 14.6 months 9.9–NE. |
|
NCT03289533 |
VEGF Liver 100 |
Avelumab + Axitinib |
AFP ≥400 ng/mL, n = 22 |
AE ORR: 13.6% (2.9–34.9) 5 DCR: 68.2 (45.1–86.1) PFS: 5.5 months (1.9–7.4) OS: 12.7 months (0.0–NE) DOR: 5.5 months (3.7–7.3) |
|
NCT03418922 |
|
Lenvatinib + Nivolumab |
first-line, (n = 30) |
DLT, AEs |
|
NCT02715531 |
GO30140 |
Atezolizumab + Bevacizumab |
n = 73 |
ORR: 27% 6 PFS: 7.5 months (0.4–23.9+) |
|
NCT01658878 |
CheckMate 040 |
Cabozantinib + Nivolumab ± Ipilimumab |
first or second-line, (dose-escalation, dose-expansion), (n = 1097, across all cohorts) |
safety, tolerability, ORR |
|
NCT03170960 |
COSMIC-021 |
Cabozantinib + Atezolizumab |
first-line, (dose-escalation and dose-expansion), (n = 1732, across all cohorts) |
MTD, ORR |
|
NCT03347292 |
|
Regorafenib + Pembrolizumab |
first-line, (dose-escalation and dose-expansion, n = 57) |
TEAE, DLT |
|
NCT03539822 |
CAMILLA |
Cabozantinib + Durvalumab |
second-line, (n = 30) |
MTD |
|
NCT03475953 |
REGOMUNE |
Regorafenib + Avelumab |
Second-line, (n = 212) |
Recommended phase II dose, ORR |
|
NCT02572687 |
|
Ramucirumab + Durvalumab |
Second-line and AFP ≥1.5x ULN, n = 28 |
DLTs ORR: 11% 7 PFS: 4.4 months (1.6–5.7) OS: 10.8 months (5.1–18.4) |
|
NCT3463876 |
RESCUE |
SHR-121 (Camrelizumab) + Apatinib |
n = 18 (n = 40) |
ORR: 38.9% 8 DCR: 83.3% PFS: 7.2 months (2.6–NE) |
|
NCT02423343 |
|
Galunisertib (TGFβ receptor I inhibitor) + Nivolumab |
second-line and AFP ≥200 ng/mL, (dose escalation and cohort expansion, n = 75) |
MTD |
|
Phase III |
|
|
|
|
|
NCT03847428 |
EMERALD-2 |
Durvalumab ± Bevacizumab versus placebo |
adjuvant, randomized, double-blinded, (n = 888) |
RFS |
|
NCT03434379 |
IMbrave150 |
Atezolizumab + Bevacizumab versus sorafenib |
first-line, randomized, open label, n = 501 |
OS: not reached for Atezolizumab + bevacizumab vs 13.2 months for sorafenib; HR 0.58, p = 0.006 9 PFS: 6.8 months for Atezolizumab + bevacizumab versus 4.3 months for sorafenib; HR 0.59, p < 0.0001 ORR: 27% |
|
NCT03713593 |
LEAP-002 |
Lenvatinib + Pembrolizumab versus Lenvatinib |
first-line, randomized, double-blinded, (n = 750) |
OS, PFS |
|
NCT03755791 |
COSMIC-312 |
Cabozantinib + Atezolizumab versus Sorafenib versus Cabozantinib |
first-line, randomized, open label, (n = 740) |
OS, PFS |
1 Bold denotes immune checkpoint inhibitors. 2 n, number of the patients analyzed in the study. The number in the parenthesis shows the number of the planned enrollment. 3 Bold denotes the primary outcome measures of the study. Duration of responses and survival are shown as median values. The numbers in the parenthesis show 95% confidential interval. 4 Ikeda et al. The American Association for Cancer Research (AACR) annual meeting 2019 (abstract #18). 5 Mudo et al. J. Clin Oncol 2019; 37 (supplement. abstract 4072). 6 Pishvaian et al. ESMO 2018 congress (# LBA26). 7 Bang et al. J Clin Oncol 2019; 37 (supplement. abstract). 8 Xu et al. J Clin Oncol 2018; 36 (supplement. abstract 4075). 9 Cheng et al. ESMO Asia2019 congress (# LBA3). DLT, dose-limiting toxicity; MTD, maximum tolerated dose; TEAEs, treatment-emergent adverse event.
3.4. Immune Checkpoint Inhibitors of Cancer Stem Cells
As previously reported, PD-L1 is expressed in the progenitor subtype of HCCs [34,38]. We also found a significant increase of PD-L1 expression in CK19-positive and/or SALL4-positive HCCs compared to those not expressing such markers [40]. Interestingly, genetic alterations involved in the PI3K-Akt pathway are more frequently detected in PD-L1-positive tumors than in PD-L1-negative tumors [40]. Inactivation of phosphatase and tensin homolog deleted from chromosome 10 (PTEN), which is known to suppress PI3K, leads to the expression of PD-L1 in glioma [71]. More importantly, a recent report suggests that an inactivating mutation of PTEN and activating mutation of PI3KCA are associated with CK19 expression in HCC [72], where expression of PD-L1 is common. As activation of the PI3K-Akt pathway is a characteristic of cancer stem cells (CSCs) [73], genetic alterations and constitutive activation of this pathway may give rise to the overexpression of PD-L1 and induce stem cell features in HCCs. From this perspective, blockade of the PD-1/PD-L1 axis may be effective for HCC with stem cell-like characteristics, which is resistant to conventional therapies. However, we have also found that infiltration of CD8+ cells is not as prominent in PD-L1-positive HCCs with mutations in the PI3K-Akt pathway compared to those without the mutations. Constitutive activation of the PI3K-Akt pathway in HCC might induce PD-L1 expression, even in a non-inflamed background, where a lack of CD8+ T-cells could be an obstacle for sufficient action of anti-PD-1/PD-L1 monotherapy. On the other hand, it is also suggested that the PI3K-Akt pathway is frequently activated in CSCs and PI3K inhibitors preferentially target CSCs [73]. As the expression of stem cell markers in HCC is associated with PD-L1 expression and since anti-PD-1/PD-L1 antibody might also target CSCs, a dual blockade of the PD1/PD-L1 axis and PI3K-Akt pathway may be an option for treating patients with HCC showing stem cell features (Figure 1c) [74].
2.5. Current Limitation of Immune Checkpoint Inhibitors and Challenge for HCC with Lack of Immune Infiltrates
HCC patients with dense lymphocyte infiltration reportedly show a marked reduction of response rate after curative resection of tumor, suggesting that TILs are critical for anti-tumor immune response [75]. From this point of view, it is conceivable that “immune cold tumor” with lack of immune infiltrates should be refractory to ICIs [66]. Ishizuka et al. reported that loss-of-function of the RNA-editing enzyme adenosine deaminase acting on RNA (ADAR1) overcomes immune checkpoint blockade resistance caused by inactivation of antigen presentation by tumor cells [76]. This restoration of sensitivity to immunotherapy may occur without recognition of TAA by CD8+ T-cells. As ADAR1 is able to act as an oncogene and its overexpression plays a role in the carcinogenesis of HCC [77], intervention of ADAR1 activity may also be a promising approach as an effective immunotherapy in patients with HCC refractory to ICIs due to the lack of CD8+ TILs (Figure 2d).
On the other hand, results from methylome analyses of cancer tissues suggest that epigenetic alterations in HCC may affect the anti-tumor immune response. Hong et al. investigated the role of epigenetic therapy on enhancing immunotherapy responses in HCC [78]. Treatment of HCC cell lines with inhibitors of enhancer of zeste homolog 2 (EZH2) and DNA methyltransferase 1 (DNMT1) improved the induction of Th1 chemokines and HCC-related antigens upon treatment with anti-PD-L1 antibody. Furthermore, using an in vivo model, they found that the combination of PD-1/PD-L1 blockade with an epigenetic modulator improves the trafficking of CD8+ T-cells into tumor tissues and promotes tumor regression. Therefore, epigenetic modulation may reactivate the epigenetically repressed chemokine responsible for T-cell trafficking and induce neoantigens as immune targets. Thus, the combination of epigenetic therapy with ICIs might also be applicable to cases with refractory HCC (Figure 1d). Schonfeld et al. showed that polymorphism in the protein arginine methyltransferase 1 (PRMT1) was associated with protein expression and modulated the expression of PD-L1 and PL-L2 in HCC cells [79], suggesting that intervention of PRMT1 activity could also restore the response to immune checkpoint inhibitors in some patients.
For the development of biomarkers that predict the tumor response to immunotherapy, it is critical to improve the outcome of the treatment. Previous reports point out that tumors with active IFN-γ signaling show immune classes that can be candidates for immunotherapy [30,39]. In addition, expression of PD-L1 in tumor cells and tumor infiltrates (CPS) was reportedly associated with tumor response in HCC cases [48]. Detection of activating mutation in CTNNB1 should also be informative to know immune cold phenotype and lack of response to ICIs in HCC [41]. On the other hand, Feun et al. indicated that baseline plasma TGF-β level could be a predictive biomarker for the response to pembrolizumab [80], and clinical trials of combined blockade of PD-1/PD-L1 and TGF-β axis are ongoing (Table 3). Dong et al. analyzed multiple tumors of the same patients for genetic structure, neoantigens, T cell receptor repertoires, and immune infiltrates, and found that only a few tumors were under the control of immunosurveillance and the majority carry a variety of immune escape mechanisms, even in a single case [81]. From this point of view, precise analysis of immune phenotype of HCC should contribute to the establishment of personalized immunotherapy in HCC cases.
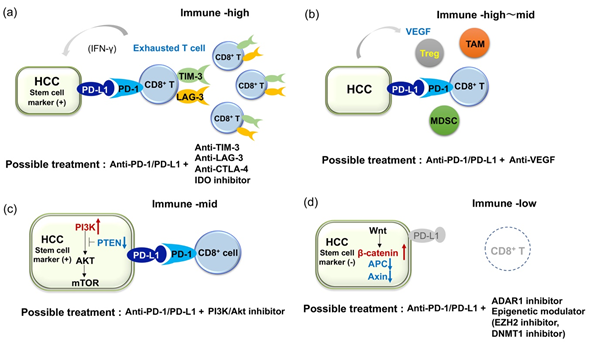
Figure 1. Illustrative figures of expected combination therapies for HCC patient refractory to immune checkpoint monotherapies. (a) In cases with expression of PD-L1 in HCC but multiple co-inhibitory receptors on tumor infiltrates, dual blockade of PD-1/PD-L1 and anti-TIM-3 or anti-LAG-3 should be required. (b) Because VEGF is known to play an important role for induction of immune suppressive molecules and cells, dual blockade of PD-1/PD-L1 and VEGF axis should be effective. (c) In cases with expression of PD-L1 and activating mutation in the PI3K-mTOR pathway in HCC, dual blockade of PD-1/PD-L1 and the PI3K-mTOR pathway might be effective. Notably, both anti-PD-1/PD-L1 and anti-PI3K-mTOR agents could target cancer stem cells (CSCs). (d) In cases with a lack of CD8+ T cell infiltration in tumor (activating mutation in the β-catenin pathway is common in this type), ADAR1 inhibitor and epigenetic modulator might induce the recruitment of CD8+ T cells into tumor and contribute to the induction of anti-tumor immunity.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12051274