Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Liposomes, nanoemulsions, solid lipid nanoparticles, nanostructured lipid nanocarriers, and lipid–polymer hybrid nanoparticles are developed for cancer treatment which is well confirmed and documented. cancer nanotechnology that overcomes the drawbacks of conventional drug delivery systems starting from small-scale barricades such as intracellular trafficking and site-specific targeting to large-scale barriers such as biodistribution.
- liposomes
- nanoemulsion
- solid lipid nanoparticles
- nanostructured lipid carriers
- lipid-polymer hybrid nanoparticles
- triple negative breast cancer
1. Lipid-Based Nanoparticles: A Versatile Drug Delivery System
Rapid advances in lipid-based nanoparticles show an immense impact on cancer treatment and management. Looking into the structural makeup of LNPs, it can be found that LNPs are composed of lipids that are both biodegradable and biocompatible such as phospholipids, cholesterol, and triglycerides. The inclusion of such less toxic substances, as well as the limited use of organic solvents, makes LNPs a safe drug delivery system against TNBC as compared to polymeric and inorganic NPs [1][2]. According to the general prototype, LNPs consists of API, lipids that are designed to sequester, deliver, and promote functionality. Further, to maintain the stability of the dispersions formed by the LNPs, in environmental stresses, surfactants or a combination of surfactants are used, depending on their HLB values [3]. Further, to safeguard the LNPs from the reticuloendothelial system (RES) and to render biological stability, LNPs are often coated with stealth or biocompatible polymeric layer such as polyethylene glycol (PEG) [4][5][6]. Finally, for possessing an enhanced targeting property, the LNPs may bind with a targeting moiety such as a biological receptor-specific ligand. Such orientation of LNPs and their small size help the LNPs to accumulate frequently on the tumor sites [4].
In cancer research, it was observed that approximately 40% of new chemical entities show poor aqueous solubility, which further limits their therapeutic efficiency. Thus, the utilization of hydrophobic LNPs as a vehicle either encapsulates or solubilizes the drug moiety that will further improve the stability of poorly aqueous soluble drugs in the surrounding aqueous media and will prevent the precipitation of the drugs both in vitro and in vivo. Hence, it could be inferred that the development of LNPs shows improved chemotherapeutic efficacy by increasing their solubility [7][8]. It was further revealed that the absorption of the poorly water-soluble chemotherapeutics in the LNPs is due to the increased solubilizing capacity of the lipids/oils. The other well-established strategies employed by LNPs in increasing the solubility of the poorly water-soluble chemotherapeutics, thereby increasing their bioavailability, including preservation of the chemotherapeutics within the lipidic matrix against chemical and enzymatic degradation, alteration in the permeability of the gastrointestinal membrane, and facilitation of the lymphatic drug transport [3].
Talking in terms of oral delivery, most chemotherapeutics are suffered from first-pass metabolism which limits their concentration in blood and subsequently at the cancer site, which restricts their therapeutic efficacy. In this context, it was found that drug delivery through the lymphatic system overturns the first-pass metabolism and improves their targeting to the cancer site. From various studies, it was observed that the triglycerides, cholesterol esters, and lipid-soluble vitamins are easily taken up by the lymphatic system and as the LNPs are mostly composed of triglycerides and arranged similarly to that of chylomicrons, the LNPs are easily taken up by the lymphatic system, thereby circumventing the first-pass metabolism, improving their targeting, and reducing toxicity. Physiologically, it was observed that the lymphatic uptake of LNPs occurs via three routes as shown in Figure 1. The first is through the gaps occurred within the lymphatic capillaries, the second is through the Peyer’s patches (Figure 1A), which are either isolated or aggregated lymphoid follicles, and the third is through the intestinal wall which follows four different mechanisms, namely, transcellular absorption, paracellular absorption, inhibition of the activity of P-glycoprotein, and cytochrome P450 and generation of chylomicrons (Figure 1B) [9].
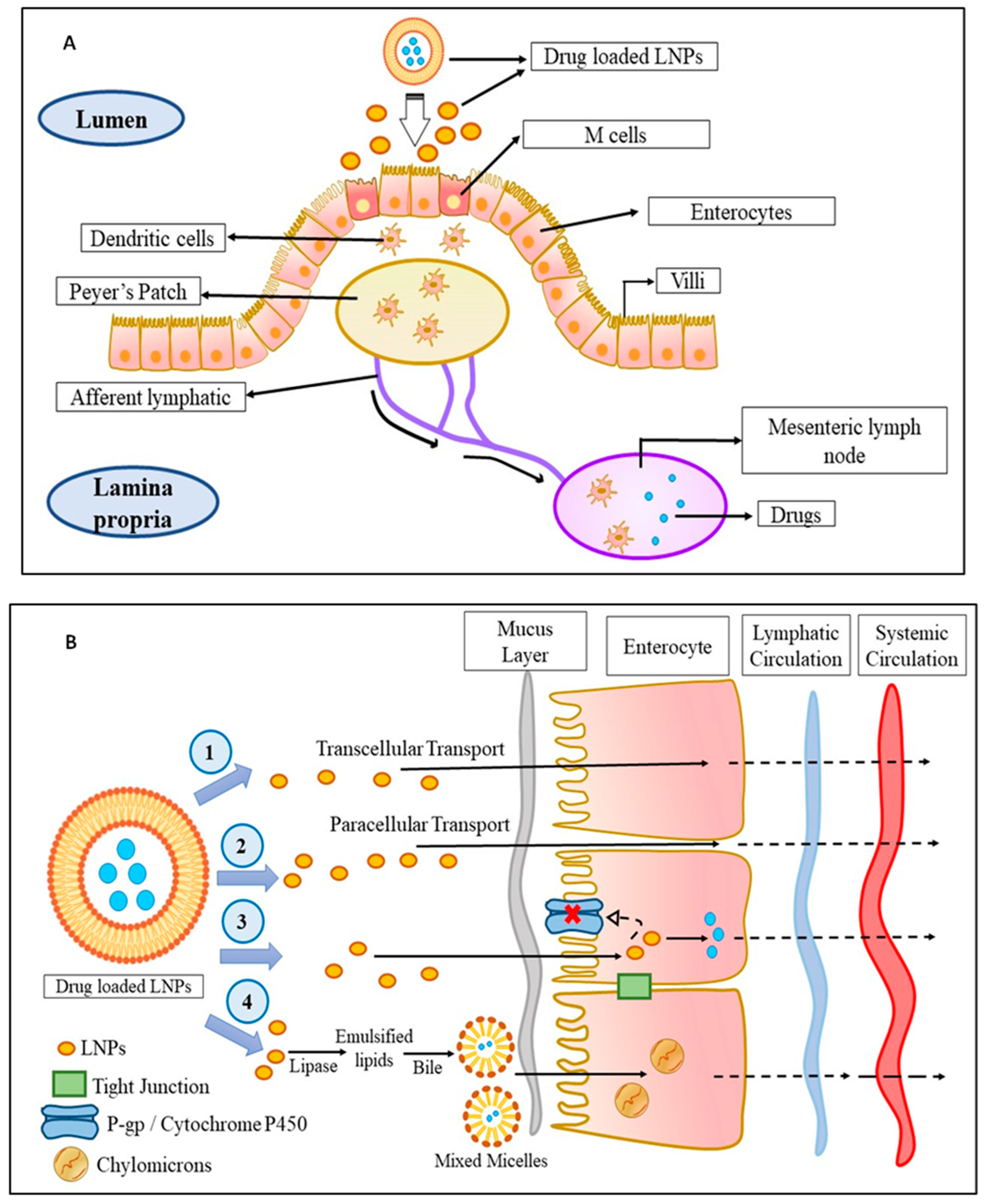
Figure 1. The mechanism of LNPs uptake into the lymphatic circulation: (A) Uptake of drug-loaded LNPs by Peyer’s patch into the lymphatic system: The drug-loaded LNPs are taken up by the M-cells of the enterocytes which are then taken up by the dendritic cells followed by Peyer’s patch from where the drug-loaded LNPs enter into the lymphatic system via afferent lymphatic. (B) Uptake of drug-loaded LNPs by an intestinal wall into the lymphatic system: The drug-loaded LNPs enter the lymphatic system through the intestinal wall in fours ways—(1) transcellular transport, (2) paracellular transport, (3) by inhibiting P-gp glycoprotein and cytochrome P450, or (4) by the production of chylomicrons. Abbreviations: M cell: membranous cell, LNPs: lipid-based nanoparticles; P-gp: P-glycoprotein.
Like cellular uptake, the in vivo fate of the LNPs also plays an important role in the drug delivery system. Basically, the entrapped drug gets released into the physiological surrounding only after breaking of the lipidic matrix. Such phenomenon occurs via lipolysis wherever lipases are found in abundance, especially in the GIT or surface erosion, in case the lipids are insensitive to lipolysis. It was observed that the LNPs composed of aliphatic esters are rapidly degraded by lipases, especially in the small intestine, while the LNPs comprised of triglycerides were first broken down by lysosomal acid lipases into diglycerides, which was then broken down into monoglycerides, and finally into fatty acids in the GIT, followed by endocytosis. Then, the lipolysates form and along with the encapsulated drugs, both are transported to the epithelial surfaces in the form on vesicles or micelles for absorption. It was further observed that apart from GIT, the lipolysis also takes place within tissues and cells. In the process of erosion, the lipid matrix undergoes either hydrolysis or dissolution which eventually plays a role in complete degradation of lipid matrices based on fatty acids and are insensitive to lipolysis. It was observed that as the chain length of the fatty acid increases, the rate of erosion of lipid matrix declines, and drug release becomes slow and steady, while the lipids with medium chain length significantly increases the erosion rate of the lipid matrix, which thereby increases the drug release from the lipid matrix [10].
It was further observed that the membrane-like structure of LNPs provides flexibility in their particle sizes which enables them to stay in the systemic circulation for a longer period by bypassing the immune responses, resulting in improved passive accumulation of LNPs in the cancer site [11]. Moreover, the particle sizes of LNPs are generally greater than 10 nm in diameter, which restricts their elimination by the kidney but allows the elimination via capillaries situated at the leaky microvasculature. Since leaky microvasculature is a common characteristic of solid tumors such as TNBC, many potent chemotherapeutics have been encapsulated in LNPs to extract the advantage of the EPR effect, which further results in increased drug accumulation at the tumor site with reduced dose regimen and systemic side effects. In this context, the encapsulation of doxorubicin (DOX) within LNPs (Doxil®/Caelyx®) increased their tumor accumulation and reduced their distribution to the myocardium, thereby decreasing the doxorubicin-induced cardiotoxicity, as compared to conventional doxorubicin solution (Adriamycin®, Pfizer, Manhattan, NY, USA) [7].
It was observed that the challenging task in the preparation of LNPs is obtaining proper size and polydispersity of the LNPs and uniform loading of chemotherapeutics within the LNPs. It was observed that the desired shape and size was obtained by controlling the process parameters employed in the various types of preparation procedure of LNPs. For instance, high pressure homogenization is one of the methods employed for the preparation of LNPs. It was observed that during hot high-pressure homogenization, very narrow particle size distribution was obtained, while in cold high-pressure homogenization, a broad size distribution was obtained. It was inferred that the size distribution of LNPs in high-pressure homogenization depends upon the temperature and pressure provided, type of homogenizer employed, and number of homogenization cycles applied. Likewise, in solvent emulsification evaporation method, the particle size of LNPs were controlled by the type and concentration of lipids, and surfactant mixture within the organic phase. It was observed that the particle size ranges between 30 and 100 nm when the lipid concentration is employed up to 5% w/v, above which the particle size increases beyond 100 nm. In case of solvent emulsification diffusion method, the particle size obtained was below 100 nm with narrow particle size distribution. It was observed that particle size increases on usage of non-ionic surfactant, while it decreases on using ionic surfactant. However, it was suggested to use a combination of two or more surfactants for better control of the particle size. Lastly, in the ultrasonication method, the particle size is obtained in the range of 30–200 nm with broad particle size distribution. It was observed that the particle size can be controlled by varying the frequency, intensity, and time of ultrasonication [12].
For the loading of the chemotherapeutics within the LNPs, the active incorporation method can be used, i.e., loading of drugs after LNPs formation, or passive i.e., loading of drugs during LNPs formation [13]. The active method involves adsorption or absorption methods that are achieved by incubating the LNPs with concentrated drug solution [14]. The passive method involves the mechanical method, solvent dispersion method, and detergent removal method [13]. It was observed that the drug loading depends upon the solubility of the drugs within the lipid matrix, which is further associated with the composition of the lipid matrix, molecular weight of the drug, the interaction between the drug and lipids and the presence of end functional groups (i.e., ester or carboxyl) in either the drug or lipid matrix [14]. LNPs were also used for the loading of nucleic acid (siRNA, mRNA, and pDNA) proteins. It was observed that fabrication of nucleic acid loaded LNPs include detergent dialysis and ethanol loading technique. However, the rapid-mixing method and T-mixing method have gained more popularity as it assures >90% entrapment efficiency. In recent times, microfluidic mixing approaches were designed based on rapid-mixing approach which further promises to fabricate nucleic acid, and protein loaded LNPs in a more reproducible and scalable fashion. It was further observed that all the mentioned methods allow rapid mixing of lipid containing organic phase into aqueous phase comprised of nucleic acid, and proteins, and resulting in an enhanced entrapment efficiency [15].
The different types of LNPs developed for the treatment of TNBC are liposomes, nanoemulsions (NEs), solid lipid nanoparticles (SLNs), nanostructured lipid carriers (NLCs), lipid–polymer hybrid nanoparticles (LPH-NPs), and exosomes (Exo). Briefly, liposomes are microscopic phospholipid bilayer nanovesicles while NEs are colloidal nanosystems with lipophilic surfaces and a negative charge. SLNs are colloidal nanosuspensions, while NLCs are colloidal blends of solid and liquid lipids. LPH-NPs are colloidal blends of lipids and polymers (non-lipid substances) [1]. Exosomes (Exo) are biological nanosized vesicles composed of lipid bilayers with embedded surface proteins [16]. The distinct characteristics, advantages, and disadvantages of each type of LNPs are mentioned in Figure 2.
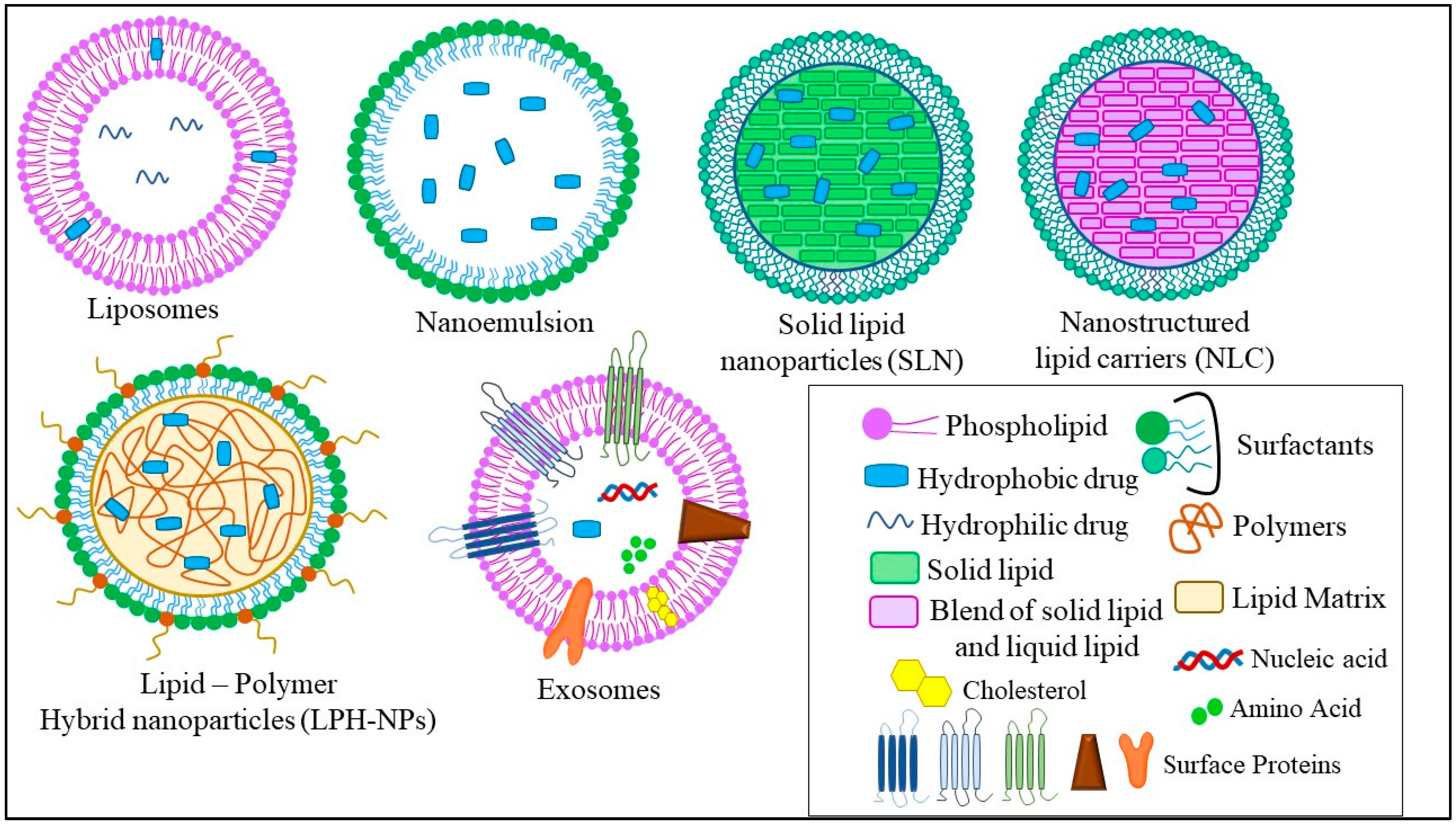
Figure 2. Different types of LNPs used for the treatment of TNBC.
2. Liposomes
Liposomes are a vesicular-type drug delivery system obtained spontaneously by dispersing the lipids (phospholipids) in aqueous media. The liposomes were first discovered in 1963 by Alec Bangham [17]. In terms of lipid shell, phospholipids form the main element of liposomes [18]. It was found that phospholipids are also considered an important constituent of the biological membrane. The phospholipids are comprised of a polar head, which is composed of hydrophilic moieties, and a non-polar tail, which is comprised of hydrophobic moieties [19][20]. Depending on the presence or absence of charges or the type of charges, liposomes are classified as uncharged, positively charged, negatively charged, and amphiphilic (zwitterionic). The positively charged lipids used in the formation of liposomes are N-[1-(2,3-dioleyloxy)propyl]-N,N,N-triethylammonium (DOTMA), and 1,2-dioleoyl-3-trimethylammoniopropane (DOTAP), while the negatively charged lipids employed in liposomes are phosphatic acid, phosphatidylserine, phosphatidylglycerol, phosphatidylinositol, and dicetylphosphate). The various zwitterionic lipids used are phosphatidylethanolamine, phosphatidylcholine, etc. [21]. It was found that the administration of the charged lipids increases the interlamellar distance between the phospholipid bilayers, which provides enhanced drug entrapment efficiency and physical stability. Further, in a study, it was observed that the presence of DMPC (dimyristoylphosphatidylcholine, a derivative of phosphatidylcholine) induced apoptosis in various cancer cell lines such as breast cancer, lung cancer, etc. [17]. Apart from phospholipids, cholesterol also exists as one of the elements of liposomes and plays a vital role in maintaining and preserving the fluidity, permeability, and stability of the phospholipids both in vitro and in vivo. Further, it was observed that using derivatives of cholesterol such as 6-aminomannose-cholesterol or glycosylated cholesteryl bypasses the RES uptake and increases the targetability of the liposomes towards cancer cells [17]. It was further found that the liposome also improves the aqueous solubility of poorly water-soluble drugs [7].
Therefore, from the above reports, it was inferred that the stability of the phospholipid bilayer, the entrapment efficiency, and the drug loading of the liposomes as well as their tissue distribution and renal clearance ultimately depend on the composition of the lipid membranes and the content of the cholesterol.
Various poorly water-soluble drugs have been administered using liposomes as a delivery system such as indomethacin, amphotericin B, and azidothymidine, which have already reached the commercial market [7].
Guo et al., 2019 developed a dual complementary liposome (DCL) composed of lipids such as 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy (polyethylene glycol)-2000] (DSPE-PEG-COOH), encapsulating doxorubicin and further surface-functionalized with antibodies against intercellular adhesion molecule–1 (ICAM1) and epithelial growth factor receptor (EGFR) for effective treatment of TNBC. It was observed that DCLs showed an average particle size of 130 ± 30 nm with a zeta potential of −6 and −10 mV. Further, it was revealed that DCLs showed enhanced internalization to MDA-MB-231 cells and MDA-MB-436 cells (42.7% and 60.9% respectively), along with a significant reduction of proliferation in vitro (30–40%). Moreover, the cancer cells (MDA-MB-231 and MDA-MB-436) treated with DCLs showed a reduction in their cell count by 64% and 46%, respectively. DCLs displayed enhanced tumor targetability and antitumor efficacy with reduced lung metastasis, depicting that the DCLs could be served as an effective therapeutic nanoplatform against TNBC [22]. Yan et al., 2019 fabricated tLyp-1-peptide modified liposomes composed of DSPE-PEG2000. The modified liposomes were prepared to encapsulate the miRNA responsible for silencing the slug gene. It was found from the previous studies that the slug gene is responsible for activating the TGF-β1/Smad pathway, causing invasion and proliferation of TNBC cells. It was found that the modified miRNA liposomes showed a particle size of 120 nm and exhibited an enhanced cellular uptake by TNBC cells in vitro, targeting mitochondria. Moreover, the modified miRNA liposomes showed enhanced anticancer activity and silenced the expression of the slug gene. In addition to these, the modified miRNA liposomes showed increased internalization to TNBC cells (48.79 ± 0.42), as compared to free miRNA complexes (3.69 ± 0.08). In addition, modified miRNA liposomes showed increased inhibitory rates (64.33 ± 8.18%), as compared to free miRNA complexes [23]. Chen et al., 2021 prepared detachable immune liposomes (ILips) as an immunochemotherapeutic approach for delivering paclitaxel and anti-CD47 in the TNBC. The lipids used for the preparation of ILips include DOPE (dioleoylphosphatidylethanolamine). It was observed that the ILips facilitate the release of CD-47 in response to MMP2 eventually polarized the M2 phenotype to M1 macrophage enhancing phagocytosis of TNBC cells and activating the responses of the T cell immune system. Paclitaxel and CD-47 showed a synergistic anticancer effect along with reduced metastasis, compared to paclitaxel liposomes and free CD-47 (2.3- and 3.1-fold, respectively). Furthermore, a lower IC50 was observed in the case of ILips as compared to paclitaxel-liposomes and free paclitaxel (2.8- and 6.4-fold respectively), which indicated that the ILips showed significant inhibition of TNBC cell proliferation. In addition to these, ILips showed an increased expression of CD80 (1.5-fold) as compared to free CD-47, indicating that the binding of PTX and CD-47 led to the effective delivery of CD47 to cancer sites along with increased polarization of macrophages [24]. Alawak et al., 2021 engineered thermoresponsive liposomes encapsulating doxorubicin for the effective treatment of TNBC. The engineered thermoresponsive liposomes were further surface-functionalized by linking with the MAB1031 antibody via covalent coupling (LipTS–GD–MAB). It was observed that the MAB1031 antibody was employed to target ADAM8, found to be overexpressed in TNBC patients. The lipids used for the fabrication of liposomes include DPPC, DSPC, cholesterol, and DSPE. The cellular toxicity study revealed that 80% of cells were found viable when the cells were treated with LipTS–GD–MAB. In addition to this, the LipTS–GD–MAB showed increased cellular internalization as compared to doxorubicin liposomes [25]. El-Senduny et al., 2021 prepared Azadiradione-loaded liposomes (AZD-lipo) for effective treatment of TNBC. It was observed that the AZD-lipo showed enhanced anti-cancer activity along with increased oral bioavailability as compared to free AZD. In addition, AZD-lipo showed less expression of proteins responsible for the proliferation of TNBC cells, and angiogenesis in TNBC cells such as cyclin D1, COX-2 (0.024 ± 0.005 at 50 μM), survivin, and VEGF-A (0.302 ± 0.01 at 25 μM) as compared to free AZD, where the concentration of COX-2 is 0.553 ± 0.015 at 50 μM, and that of VEGF-A is 0.801 ± 0.011 at 25 μM. In addition, the AZD-lipo showed decreased IC50 values (26.85 ± 3.48 μM), as compared to free AZD (44.88 ± 2.57 μM). Such observations indicated an increased bioavailability of AZD in the biological system from AZD-lipo. Hence, it could be inferred that liposomes provide an effective therapeutic strategy for increased delivery of low bioavailable drugs and effective treatment against TNBC [26].
3. Nanoemulsions (NEs)
Nanoemulsions (NEs) are an isotropic thermodynamically stable system composed of two immiscible liquids that are equilibrated into a monophase using surfactants or a mixture of surfactants and co-surfactants. The nanoemulsions are composed of oils, surfactants, or their mixture and aqueous phase. The oil phase is used as a solubilizer for hydrophobic drugs [27]. For the delivery of the hydrophobic drugs via NE, the drugs are incorporated into the oil phase to form nanodroplets dispersed into the continuous aqueous phase and provide an oil in water (O/W) NE system [28]. It was further observed that the rate of drug release from the NE primarily depends on the oil/water partition coefficient of the drug molecule, oil/lipidic content as well as the water content of the NE system [7]. In NE, 5–20 wt% of the oil or lipid is considered as the dispersed phase. Hence, screening of oil or lipids is proven to be an important aspect of nanoemulsion formation, as the API must get freely solubilized in the oils/lipids before their dispersion to the aqueous phase. The various oils/lipids used for the lipid phase are glycerides, medium-chain triglycerides, long-chain unsaturated fatty acids, vegetable oils, and polyalcohol esters of medium-chain fatty acids [29]. It was observed from various studies that the nanoemulsions developed using long-chain triglycerides (LCT) showed an average particle size of 120 nm, while those prepared using short-chain triglycerides (SCT) showed smaller particle sizes (40 nm), compared to the former ones. In addition, the lipidic core of the nanoemulsion exhibits an impact on drug loading, stability, and physicochemical attributes. The surfactant or the mixture of surfactants and co-surfactants is employed to reduce the interfacial tension between the lipid phase and aqueous phase for the development of a thermodynamically stable monophasic system. Moreover, it was found apart from reducing the surface tension, a suitable surfactant stabilizes the interfacial surface via electrostatic interactions. The various emulsifying agents used in nanoemulsion preparation include surfactants such as Tween 80, sodium dodecyl sulfate, phospholipids such as soy lecithin, zwitterionic proteins such as caseinate, polysaccharides such as modified starch, and polymers such as PEGs [30]. Various studies showed that the small size, large surface area, and tunable surface characteristics help the nanoemulsion in increasing their circulation half-life, and specific targetability towards cancer sites. As cancer cells are fenced by leaky vasculature, the nanoemulsions can easily bypass the physiological barriers and accumulate within the cancer cells [31]. Various anticancer drugs such as tamoxifen and dacarbazine have been administered in the NE system [7].
Kim et al., 2019 developed decitabine (DAC)- and panobinostat (PAN)-loaded nanoemulsion which was further coated with lysophophatidylcholine and lysophophatidic acid for targeting LPC receptor and LPAR1 receptor that is overexpressed in TNBC cells. It was observed that the DAC-PAN-LNEs restored CDH1/E-cadherin and suppressed the expression of FOXM1 which eventually inhibited the growth of TNBC cells. Further, it was observed that DAC/PAN LNEs decreased the cell viability of MDA-MB-231 by 55% which indicated the increased therapeutic activity of NEs against TNBC. In addition, DAC/PAN-LNEs synergistically decreased the expression of FOXM1 mRNA and FOXM1 protein expressions by 80% [32]. Xu et al., 2020 developed puerarin nanoemulsion (NanoPue) using soya lecithin and Kolliphor® HS15 for increased oral bioavailability and therapeutic efficacy against TNBC. It was observed that the NanoPue reduced the expression of tumor-associated fibroblast (TAFs) and enhanced the intra-tumoral infiltrations (ITLs) of cytotoxic T cells by 6-fold and 2-fold, respectively, as compared to control, and mediated chemotherapy effect of nano-paclitaxel in the desmoplastic triple-negative breast cancer (TNBC) model. Such an activated immune-microenvironment caused by NEs treatment facilitated a synergistic PD-L1 blockage approach for the treatment of TNBC [33]. Han et al., 2021 fabricated elemene nanoemulsion (E-NE) for the treatment of TNBC as well as to inhibit their metastasis to the lung. The lipidic phase is comprised of soybean phospholipids and cholesterol. It was observed that E-NE reduced the stabilization of HIF-1α by effectively scavenging ROS. Additionally, the E-NE limited angiogenesis and NLRP3 inflammasomes and IL-1β [34]. Saraiva et al., 2021 developed edelfosine nanoemulsion (ET-NEs) containing lipids such as miglyol 812 and phosphatidylcholine. It was observed that the ET-NEs decreased tumor growth in vitro and in vivo. Furthermore, it was observed that the ET-NEs penetrated the physiological barriers of MDA-MB 231 xenografted zebrafish embryos, resulting in a significant reduction of cancer cell proliferation, which was further confirmed by confocal laser microscopy. Further, it was observed that the ET-NEs showed a dose-dependent IC50 which was found to be 6.9 μg/mL at 13.2 μM after 24 h of incubation, whereas the free ET showed a higher IC50 which is 13.9 μg/mL at 26.5 μM [35].
4. Solid Lipid Nanoparticles (SLNs)
Solid lipid nanoparticles (SLNs) are O/W type of colloidal nanoparticles consisting of an inner lipid-based phase and outer aqueous-based phase that are stabilized by surfactants or a mixture of surfactants [36][37]. It was observed from various studies that SLNs can either solubilize lipophilic chemotherapeutics homogeneously within the lipidic matrix or develop a drug-enriched shell surrounding the lipidic core [38][39]. Further, it was observed that based on the type of drug deposition pattern within the lipid matrices, i.e., either chemotherapeutics-enriched core or chemotherapeutics-enriched shell, the drug release profile can be regulated to the advantage for the fulfillment of desired release profile [40]. For example, SLNs with drug-enriched shells exhibit a biphasic drug-release profile through initial burst release from the outer shell followed by gradual release from the lipid core. However, in SLNs with drug-enriched core, a more prolonged sustained release profile was observed due to enhanced drug diffusional distance from the lipidic core [7]. The lipid-based phase used is the solid lipids including triglycerides, fatty acids, steroids, and waxes [41]. It was further reported that the lipids and surfactants used for the preparation of SLNs should fall under GRAS (Generally Recognized as Safe) regulations. The most widely used solid lipids are palmitic acid, stearic acid (SA), glyceryl monostearate (GMS), compritol 888 ATO, trimyristin, Capmul®MCM C10, soybean lecithin, etc. In terms of structure, SLNs show similarity with emulsion except for the fact that the SLNs replaced the oily core with a lipid-based core. Moreover, the SLNs also show a certain amount of similarity as well as dissimilarity with the conventional liposomes. It was observed that like liposomes, SLNs are composed of lipids and unlike liposomes, the SLNs do not have a lipidic bilayer instead composed of a micelle-like structure. It was stated that lipids used in the SLNs should remain in solid form at room temperature and body temperature [42]. Such characteristics help in encapsulating lipophilic drugs in the melted lipid phase which further help in increasing the drug-loading capacity of SLNs and altering the physicochemical properties of drugs both in vitro and in vivo [43][44]. Such features also aid in reducing the degradation profile of the lipids making them suitable for the fabrication of the controlled release formulation. Rheologically, it was observed that the formation of SLNs depends on the interfacial tension (adhesive forces) between the two phases, where the addition of one or more surfactants the interfacial tension by reducing the surface energy and facilitates the formation of stable SLNs [43]. From various studies, it was observed that typically, SLNs are comprised of 0.1–30% solid lipid and 0.5–30% surfactant or surfactant blend [45]. SLNs exhibit increased loading capacity, entrapment efficiency, and less toxicity as compared to polymeric nanoparticles. Therapeutically, SLNs show enhanced targetability to cancer cells via passive targeting as well as active targeting with some external modifications. Additionally, SLNs could encapsulate various moieties such as drugs, proteins, nucleic acids, etc., and improve their pharmacokinetic attributes and physicochemical stability [41]. SLNs have been fabricated for the delivery of various lipophilic chemotherapeutics such as camptothecin, all-trans retinoic acid, etc. Despite the advantages provided by the SLNs, they do exhibit certain physical instability upon storage. It was observed that the solid lipids undergo crystallization during storage which limits the movement of active moieties within the lipidic core, mediating an expulsion of active moieties into the dispersion media and affecting the entrapment efficiency of the lipid-based nanosystem [36].
Eskiler et al., 2018 prepared BMN-673 loaded SLNs using GMS as solid lipids and Tween 80 as a surfactant to improve its therapeutic index and to overcome the BRCA1 mutated sensitive and resistant TNBC. It was observed that compared to native BMN 673, BMN 673-SLNs showed a significant decrease in HCC1937 and HCC1937-R cells with less damage to TNBC cells. In addition, BMN 673-SLNs induced significant toxicity in TNBC cells via breaking of double-stranded DNA, arresting of G2/M cell cycle, and cleaving of PARP moieties [46]. Siddhartha et al., 2018 developed di-allyl-disulfide (DADS)-loaded solid lipid nanoparticles using palmitic acid as solid lipid and pluronic F-68 and soy lecithin as surfactant mix, which was further conjugated with RAGE antibody to enhance the targetability and delivery of DADS to TNBC cells. It was observed that the DADS-RAGE-SLNs significantly increased the cytotoxicity and apoptosis (61.8%) as compared to DADS (15%). Additionally, DADS-RAGE-SLNs showed enhanced cellular internalization via receptor-mediated endocytosis as the SLNs bypassed P-gp efflux proteins as compared to DADS [47]. Kothari et al., 2019 fabricated docetaxel (DTX)–alpha-lipoic acid (ALA) co-loaded SLNs using GMS, SA, and Compritol ATO 888 as solid lipids and Tween 80 as a surfactant to treat TNBC. It was observed that the DTX-ALA-SLNs showed increased cytotoxicity to 4T1 cells as compared to DTX-SLNs, ALA-SLNs, and free drugs. Moreover, the DTX-ALA SLNs showed increased apoptosis of 32% as compared to free DTX which is only 11% [48]. Pindiprolu et al., 2019 prepared niclosamide-loaded SLNs (Niclo-SLNs) using stearyl amine as solid lipid and Tween 80, and pluronic F-68 as surfactant mix for the treatment of TNBC. It was observed that the Niclo-SLNs showed increased cytotoxicity and enhanced cellular internalization (77.06%) at the G0/G1 phase of the cell cycle as compared to free Niclo (69.50%). It was inferred that Niclo-SLNs showed increased cellular uptake due to their ability to bypass the efflux pump and increased absorption of drugs within the cancer cells [49]. In this context, Pindiprolu et al., 2020 fabricated phenylboronic acid-modified Niclo-SLNs (PBA-Niclo-SLNs) to enhance the targetability of Niclo to TNBC cells, thereby increasing its therapeutic efficacy toward TNBC cells. It was observed that PBA-Niclo-SLNs showed increased cytotoxicity (CTC50 7.311 ± 2.1 μM), inhibition of cell proliferation at G0/G1 cell cycle (74.01 ± 0.60%) and apoptosis (21.3 ± 1.0%) as compared to Niclo-SLNs (CTC50 18.49 ± 2.5 μM; 61.01 ± 1.10%; 12.3 ± 1.1%), and free Niclo (CTC50 31.17 ± 3.2 μM; 54.21 ± 0.90%; 10.8 ± 0.9%), respectively. Additionally, PBA-Niclo-SLNs significantly inhibited STAT3, TNBC stem cell populations (CD44+/CD24−), and EMT (epithelial–mesenchymal transition) markers along with increased tumor-site accumulation with significant tumor regression and enhanced survivability of TNBC-bearing mice [50].
5. Nanostructured Lipid Carriers (NLCs)
Nanostructured lipid carriers (NLCs) are considered second-generation lipid-based nanoparticles. It is composed of a mixture of solid lipid and liquid lipid which is further stabilized in the aqueous phase via one or more surfactants [51]. The incorporation of liquid lipid and solid lipid within the lipid matrix forms a massive crystal imperfection or amorphous structure that facilitates enhanced loading of drugs into the lipid matrix with less pronounced drug expulsion [45]. It was further observed that the NLCs obtained should remain solid at a temperature higher than 40 °C. Generally, NLCs encapsulate approximately 5% of drug w/v where approximately 3 to 4% drug loading is obtained (entrapment efficiency of ≈70%) [45]. The various solid lipids employed in NLCs preparation are glyceryl tripalmitate, softisan 154, glyceryl monostearate, compritol ATO 888, stearic acid, precirol, PEG-DSPE, soybean phosphatidylcholine, etc., and the liquid lipids used for the preparation of NLCs include glyceryl tridecanoate, olive oil, labrafil WL 2609 BS, oleic acid, labrafil M2125 Cs, labrafac PG, polyoxyl castor oil, etc. From various studies, it was observed that the NLCs show high tolerability due to the existence of lipids that are bio-compatible and tunable. Such characteristics enable NLCs to show increased drug loading capacity, decreased risk of gelation, and restricted leakage of the drug upon storage. In addition to these, NLCs also extend their exposure period over tumor cells via the EPR effect, thereby increasing the therapeutic efficacy of the antitumor drug on the tumor site. The NLCs are further multi-functionalized to increase the drug payload, increase targetability to the cancer site, and release the drug in a more controlled way [45].
Pedro et al., 2019 prepared paclitaxel-loaded NLCs (PTX-NLCs) using compritol ATO 888 as solid lipid and MCT as liquid lipid to increase its therapeutic efficacy against TNBC. The NLCs were further stabilized by using Tween 80 and soya lecithin. It was observed that the PTX-NLCs showed increased in-vitro cell cytotoxicity and anti-clonogenic activity against MDA-MB-231 cells as compared to free PTX. Further, from the cell viability assay, it was observed that the free PTX showed more cell viability which is 56.0 ± 3.2% as compared to PTX-NLCs (38.0 ± 5.0%). Additionally, PTX-NLCs exhibited 1.5- and 1.7-fold increased tumor site accumulation after 30 and 120 min, respectively, in tumor-bearing mice, as compared to free PTX [52]. Zhang et al., 2019 fabricated folic acid (FA)-functionalized paclitaxel (PTX) and chlorin e6 (Ce6)-loaded NLC (PTX-Ce6-NLC) to increase their targetability and therapeutic efficacy against TNBC. The NLCs were prepared using Precirol ATO 5 as solid lipid and Maisine 35-1 as liquid lipid, stabilized by Cremophor RH40. It was observed that FA-PTX-Ce6-NLC showed enhanced MDA-MB-231 cellular uptake via FR-mediated endocytosis as compared to free PTX. Moreover, it was observed that Ce6 dissociated and evenly distributed in tumor cells. Additionally, from the pharmacodynamic study, it was observed that the NLCs showed enhanced drug-loading without side effects as compared to free PTX [53].
Lages et al., 2020 developed doxorubicin- and α-tocopherol succinate-loaded NLCs using compritol 888 ATO as solid lipid and docosahexaenoic acid (DHA) as liquid lipid to increase their anti-cancer activity against TNBC. Tween 80 was employed as a surfactant to further stabilize the lipid phased in aqueous media. It was observed that the NLCs showed a controlled release profile with an increased release in acidic media. Further, the NLCs showed decreased mortality in mice, reduced metastasis to lungs, and prevented drug-induced toxicity to vital organs (heart and liver) as observed from biochemical and histological assays. In addition, the NLCs showed a higher tumor inhibition ratio (76.6%) as compared to free doxorubicin (64.6%) [54]. Gadag et al., 2021 prepared resveratrol-loaded NLCs (RVT-NLC) using GMS as solid lipid and caproyl 90 as liquid lipid for increasing the therapeutic efficacy of resveratrol against TNBC. The NLCs were stabilized using labrasol as a surfactant. From the cell viability study, it was observed that the RVT-NLCs showed decreased MDA-MB-231 cell-viability (IC50 = 27.50 ± 3.43 μg/mL), as compared to free RVT (IC50 = 33.93 ± 7.34 μg/mL), which indicated that the RVT-NLCs were found to be more potent as compared to free RVT. Further, to increase the therapeutic efficacy via the dermal route, the RVT-NLCs were loaded within microneedle. It was observed that the RVT-NLCs loaded microneedle showed increased skin permeation, improved cellular internalization, and prevented metastasis as compared to free RVT. In addition, the RVT-NLCs increased pharmacokinetic attributes (Cmax = 343.75 ± 31.89 ng/mL; AUC0-t = 4529.2 ± 299.67 h∗ng/mL), as compared to free RVT (Cmax = 269.30 ± 30.26 ng/mL; AUC0-t = 458.3 ± 21.21 h∗ng/mL) [55]. Gilani et al., 2021 prepared luteolin-loaded NLCs (LTN-NLC) using GMS as solid lipid and caproyl 90 as liquid lipid to treat TNBC, stabilized by poloxamer 188. Further, to obtain a sustained release profile, the NLCs were surface functionalized by chitosan (LTN-CS-NLCs). It was observed that LTN-CS-NLCs exhibited a slow-release profile of LTN during a 24 h study. Moreover, LTN-CS-NLCs showed increased mucoadhesion, improved gastrointestinal stability, and intestinal permeation as compared to free LTN. In addition, from the MTT assay, it was observed that LTN-CS-NLCs showed decreased MDA-MB-231 cell viability (IC50 = 11.48 ± 2.38 μM), as compared to free LTN (IC50 = 29.64 ± 3.84 μM) after 48 h treatment. Additionally, LTN-CS-NLCs exhibited 4.3-fold increased intestinal permeation as compared to LTN suspension, indicating the superiority of NLCs in overcoming P-gp efflux pump-mediating elimination, in comparison to suspension [56].
6. Lipid Polymer Hybrid Nanoparticles (LPH-NPs)
Lipid–polymer hybrid nanoparticles (LPH-NPs) are considered new-generation nanoparticles exploiting the advantages of both polymeric nanoparticles and lipid-based nanoparticles in a single nanosystem [57]. Such a system is comprised of a polymeric core surrounded by a lipidic monolayer, which could be further surface-functionalized via PEGylation to prolong its circulation or via ligand to enhance its targetability. LPH-NPs impart certain characteristics of lipid-based nanoparticles which include enhanced drug-loading capacity, biodegradable and biomimetic nature, and certain traits of polymeric nanoparticles such as controlled/sustained drug release profile and a variety of surface functionalization or modification. The various polymers used in the preparation of LPH-NPs are approved by the Food and Drug Administration (FDA) which include polycaprolactone (PCL), poly (lactic-co-glycolic acid) (PLGA), polylactic acid (PLA), poly β-amino ester (PbAE), etc. In terms of lipidic components, charged or zwitterionic lipids are selected as they could be exploited to mediate covalent or non-covalent bonds with the desired ligands, antibodies, and nucleic acids (DNA, RNA), proteins, or peptides. Moreover, the presence of charged moieties helps to facilitate electrostatic interaction between lipids and oppositely charged polymers (core), which would result in the development of self-assembling nanostructures. The various lipids used are DOTAP/DOPE, stearic acid, cholesterol, lecithin, lipoid GmbH, DSPE-PEG, and PEG2000-Mal [58]. It was further observed that the lipidic monolayer acts as a molecular barricade that alleviates the loss of loaded drugs throughout the LPH-NP preparation and safeguards the polymeric core from deterioration by inhibiting the diffusion of water into the polymeric core. Due to the hybrid nanostructure, LPH-NPs can incorporate anticancer drugs of different physicochemical profiles. They are also able to conjugate ligands that are overexpressed on cancer cells, resulting in enhanced targetability and therapeutic efficacy with restricted off-site toxicities. In addition, LPH-NPs exhibit improved mechanical stability upon storage. Further, it was observed that the LPH-NPs of dimensions ≤ 100 nm display increased intra-cellular release of drug(s), resulting in decreased cytotoxicity.
Zhang et al., 2017 prepared RGD-conjugated doxorubicin (DOX) and mitomycin C (MMC) co-loaded lipid–polymer hybrid nanoparticles (DMPLN) to treat TNBC. In the preparation of DMPLN, HPESO (hydrolyzed polymer of epoxidized soyabean oil) was used as a polymeric core and myristic acid was used as a lipid layer. It was observed that the RGD-DMPLN facilitated certain morphological changes and induced cytotoxicity. Moreover, compared to free drugs, RGD-DMPLN showed increased cellular accumulation, restricted lung metastasis (31-fold), remarkably decreased toxicity to the liver and heart, and improved median survival time (57%) [59]. Zhou et al., 2017 formulated calcium-phosphate-based lipid–polymer hybrid nanoparticles (LPH-NPs) co-loaded with paclitaxel and inhibitors of microRNA-221/222 for the treatment of TNBC. The LPH-NPs were prepared by using PLGA and PEG as polymers and dioleoylphosphatidic acid (DOPA) as anionic lipid. It was observed that the cell viability got decreased by approximately 80% in the case of co-loaded LPH-NPs as compared to free paclitaxel at the same dose of 0.67 μg/mL. Additionally, the co-loaded LPH-NPs showed enhanced intracellular activity as compared to free paclitaxel [60]. Garg et al., 2017 prepared fucose-anchored lipid–polymer hybrid nanoparticles (LPH-NPs) co-loaded with methotrexate (MTX) and aceclofenac (ACL) for the treatment of TNBC. LPH-NPs were prepared using gelucire 48/16 (lipid layer), phospholipid 90NG, and phospholipid S100 (polymer layer). Further, the LPH-NPs were conjugated with DSPE-PEG (2000)-NH-gructose. It was observed that LPH-NPs exhibited rapid MDA-MB-231 cellular internalization within 2 h and showed 10-fold increased bioavailability as compared to free drugs. Additionally, LPH-NPs showed ~21–25% less MDA-MB-231 cell growth, and 5–6 times increased mean residence time (MRT) as compared to free drugs [61]. Bakar-Ates et al., 2020 developed cucurbitacin B-loaded lipid polymer hybrid nanoparticles (CuB-NPs) by using polymers such as PLGA, DSPE-PEG, and lipids such as lecithin for the treatment of TNBC by inducing apoptosis in MDA-MB-231 cells. It was observed that the CuB-NPs showed decreased cell viability at 0.1 and 5 μM concentrations as compared to the control. Additionally, the treatment with CuB-NPs showed increased cell population at G0/G1 phase (56.50 ± 1.23%), and apoptosis (20.66 ± 1.99%) as compared to free CuB (47.20 ± 1.02% and; 3.69 ± 0.57%, respectively) [62].
This entry is adapted from the peer-reviewed paper 10.3390/ijms231710068
References
- Souhaila H. El Moukhtari; Carlos Rodríguez-Nogales; María J. Blanco-Prieto; Oral lipid nanomedicines: Current status and future perspectives in cancer treatment. Advanced Drug Delivery Reviews 2021, 173, 238-251, 10.1016/j.addr.2021.03.004.
- Siddartha Venkata Talluri; Gowthamarajan Kuppusamy; Veera Venkata Satyanarayana Reddy Karri; Shashank Tummala; Subbarao Madhunapantula; Lipid-based nanocarriers for breast cancer treatment – comprehensive review. Drug Delivery 2015, 23, 1291-1305, 10.3109/10717544.2015.1092183.
- Katja Cerpnjak; Alenka Zvonar; Mirjana Gašperlin; Franc Vrečer; Lipid-based systems as a promising approach for enhancing the bioavailability of poorly water-soluble drugs. Acta Pharmaceutica 2013, 63, 427-445, 10.2478/acph-2013-0040.
- Andrew D. Miller; Lipid-Based Nanoparticles in Cancer Diagnosis and Therapy. Journal of Drug Delivery 2013, 2013, 1-9, 10.1155/2013/165981.
- Varun Kushwah; Sameer S. Katiyar; Ashish Kumar Agrawal; Ramesh C. Gupta; Sanyog Jain; Co-delivery of docetaxel and gemcitabine using PEGylated self-assembled stealth nanoparticles for improved breast cancer therapy. Nanomedicine: Nanotechnology, Biology and Medicine 2018, 14, 1629-1641, 10.1016/j.nano.2018.04.009.
- Varun Kushwah; Sameer S. Katiyar; Ashish Kumar Agrawal; Isha Saraf; Inder Pal Singh; Dimitrios A. Lamprou; Ramesh C. Gupta; Sanyog Jain; Implication of linker length on cell cytotoxicity, pharmacokinetic and toxicity profile of gemcitabine-docetaxel combinatorial dual drug conjugate. International Journal of Pharmaceutics 2018, 548, 357-374, 10.1016/j.ijpharm.2018.07.016.
- Sok Bee Lim; Amrita Banerjee; Hayat Önyüksel; Improvement of drug safety by the use of lipid-based nanocarriers. Journal of Controlled Release 2012, 163, 34-45, 10.1016/j.jconrel.2012.06.002.
- Palei, N.N.; Mohanta, B.C.; Sabapathi, M.L.; Das, M.K. Lipid-based nanoparticles for cancer diagnosis and therapy; Elsevier Inc.: Cambridge, MA, USA, 2018; pp. 415–470.
- Yusrida Darwis; Arshad Ali Khan; Jahanzeb Mudassir; Noratiqah Mohtar; Advanced drug delivery to the lymphatic system: lipid-based nanoformulations. International Journal of Nanomedicine 2013, 8, 2733-44, 10.2147/ijn.s41521.
- Jianping Qi; Jie Zhuang; Yi Lu; Xiaochun Dong; Weili Zhao; Wei Wu; In vivo fate of lipid-based nanoparticles. Drug Discovery Today 2016, 22, 166-172, 10.1016/j.drudis.2016.09.024.
- Min Woo Kim; Seung-Hae Kwon; Jung Hoon Choi; Aeju Lee; A Promising Biocompatible Platform: Lipid-Based and Bio-Inspired Smart Drug Delivery Systems for Cancer Therapy. International Journal of Molecular Sciences 2018, 19, 3859, 10.3390/ijms19123859.
- Rishi Paliwal; R. Jayachandra Babu; Srinath Palakurthi; Nanomedicine Scale-up Technologies: Feasibilities and Challenges. AAPS PharmSciTech 2014, 15, 1527-1534, 10.1208/s12249-014-0177-9.
- Jayanta Kumar Patra; Gitishree Das; Leonardo Fernandes Fraceto; Estefania Vangelie Ramos Campos; Maria Del Pilar Rodriguez-Torres; Laura Susana Acosta-Torres; Luis Armando Diaz-Torres; Renato Grillo; Mallappa Kumara Swamy; Shivesh Sharma; et al. Nano based drug delivery systems: recent developments and future prospects. Journal of Nanobiotechnology 2018, 16, 1-33, 10.1186/s12951-018-0392-8.
- Rajesh Singh; James W. Lillard; Nanoparticle-based targeted drug delivery. Experimental and Molecular Pathology 2009, 86, 215-223, 10.1016/j.yexmp.2008.12.004.
- Bárbara B. Mendes; João Conniot; Aviram Avital; Dongbao Yao; Xingya Jiang; Xiang Zhou; Noga Sharf-Pauker; Yuling Xiao; Omer Adir; Haojun Liang; et al. Nanodelivery of nucleic acids. Nature Reviews Methods Primers 2022, 2, 1-21, 10.1038/s43586-022-00104-y.
- Dulla Naveen Kumar; Aiswarya Chaudhuri; Farrukh Aqil; Deepa Dehari; Radha Munagala; Sanjay Singh; Ramesh C. Gupta; Ashish Kumar Agrawal; Exosomes as Emerging Drug Delivery and Diagnostic Modality for Breast Cancer: Recent Advances in Isolation and Application. Cancers 2022, 14, 1435, 10.3390/cancers14061435.
- J. L. Arias; B. Clares; M. E. Morales; V. Gallardo; M. A. Ruiz; Lipid-based drug delivery systems for cancer treatment.. Current Drug Targets 2011, 12, 1151-1165, 10.2174/138945011795906570.
- Varun Kushwah; Devesh Kumar Jain; Ashish Kumar Agrawal; Sanyog Jain; Improved antitumor efficacy and reduced toxicity of docetaxel using anacardic acid functionalized stealth liposomes. Colloids and Surfaces B: Biointerfaces 2018, 172, 213-223, 10.1016/j.colsurfb.2018.08.047.
- Sanyog Jain; Harshad Harde; Anura Indulkar; Ashish Agrawal; Improved stability and immunological potential of tetanus toxoid containing surface engineered bilosomes following oral administration. Nanomedicine: Nanotechnology, Biology and Medicine 2014, 10, 431-440, 10.1016/j.nano.2013.08.012.
- Sanyog Jain; Swapnil R. Patil; Nitin K. Swarnakar; Ashish K. Agrawal; Oral Delivery of Doxorubicin Using Novel Polyelectrolyte-Stabilized Liposomes (Layersomes). Molecular Pharmaceutics 2012, 9, 2626-2635, 10.1021/mp300202c.
- Temidayo O. B. Olusanya; Rita Rushdi Haj Ahmad; Daniel M. Ibegbu; James R. Smith; Amal Ali Elkordy; Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules 2018, 23, 907, 10.3390/molecules23040907.
- Peng Guo; Jiang Yang; Daxing Liu; Lan Huang; Gillian Fell; Jing Huang; Marsha A. Moses; Debra T. Auguste; Dual complementary liposomes inhibit triple-negative breast tumor progression and metastasis. Science Advances 2019, 5, eaav5010, 10.1126/sciadv.aav5010.
- Yan Yan; Xue-Qi Li; Jia-Lun Duan; Chun-Jie Bao; Yi-Nuo Cui; Zhan-Bo Su; Jia-Rui Xu; Qian Luo; Ming Chen; Ying Xie; et al. Nanosized functional miRNA liposomes and application in the treatment of TNBC by silencing Slug gene. International Journal of Nanomedicine 2019, ume 14, 3645-3667, 10.2147/ijn.s207837.
- Mingshu Chen; Yunqiu Miao; Kun Qian; Xin Zhou; Linmiao Guo; Yu Qiu; Rui Wang; Yong Gan; Xinxin Zhang; Detachable Liposomes Combined Immunochemotherapy for Enhanced Triple-Negative Breast Cancer Treatment through Reprogramming of Tumor-Associated Macrophages. Nano Letters 2021, 21, 6031-6041, 10.1021/acs.nanolett.1c01210.
- Mohamad Alawak; Alice Abu Dayyih; Gihan Mahmoud; Imran Tariq; Lili Duse; Nathalie Goergen; Konrad Engelhardt; Shashank Reddy Pinnapireddy; Jarmila Jedelská; Muhannad Awak; et al. ADAM 8 as a novel target for doxorubicin delivery to TNBC cells using magnetic thermosensitive liposomes. European Journal of Pharmaceutics and Biopharmaceutics 2020, 158, 390-400, 10.1016/j.ejpb.2020.12.012.
- Fardous F. El-Senduny; Miram Altouhamy; Gamal Zayed; Choudhary Harsha; Renjitha Jalaja; Sasidhar Balappa Somappa; Mangalam S. Nair; Ajaikumar B. Kunnumakkara; Fahd M. Alsharif; Farid A. Badria; et al. Azadiradione-loaded liposomes with improved bioavailability and anticancer efficacy against triple negative breast cancer. Journal of Drug Delivery Science and Technology 2021, 65, 102665, 10.1016/j.jddst.2021.102665.
- E B Souto; A P Nayak; R S R Murthy; Lipid nanoemulsions for anti-cancer drug therapy.. Die Pharmazie 2011, 66, 473-478, .
- Sanyog Jain; Tanya Garg; Varun Kushwah; Kaushik Thanki; Ashish Kumar Agrawal; Chander Parkash Dora; α-Tocopherol as functional excipient for resveratrol and coenzyme Q10-loaded SNEDDS for improved bioavailability and prophylaxis of breast cancer. Journal of Drug Targeting 2017, 25, 554-565, 10.1080/1061186x.2017.1298603.
- Agnieszka Gawin-Mikołajewicz; Karol P. Nartowski; Aleksandra J. Dyba; Anna M. Gołkowska; Katarzyna Malec; Bożena Karolewicz; Ophthalmic Nanoemulsions: From Composition to Technological Processes and Quality Control. Molecular Pharmaceutics 2021, 18, 3719-3740, 10.1021/acs.molpharmaceut.1c00650.
- Srinivas Ganta; Meghna Talekar; Amit Singh; Timothy P. Coleman; Mansoor M. Amiji; Nanoemulsions in Translational Research—Opportunities and Challenges in Targeted Cancer Therapy. AAPS PharmSciTech 2014, 15, 694-708, 10.1208/s12249-014-0088-9.
- Elena Sánchez-López; Mariana Guerra; João Dias-Ferreira; Ana Lopez-Machado; Miren Ettcheto; Amanda Cano; Marta Espina; Antoni Camins; Maria Luisa Garcia; Eliana B. Souto; et al. Current Applications of Nanoemulsions in Cancer Therapeutics. Nanomaterials 2019, 9, 821, 10.3390/nano9060821.
- Bumjun Kim; Caroline D. Pena; Debra T. Auguste; Targeted Lipid Nanoemulsions Encapsulating Epigenetic Drugs Exhibit Selective Cytotoxicity on CDH1–/FOXM1+ Triple Negative Breast Cancer Cells. Molecular Pharmaceutics 2019, 16, 1813-1826, 10.1021/acs.molpharmaceut.8b01065.
- Huan Xu; Mengying Hu; Mengrui Liu; Sai An; Kaiyun Guan; Menglin Wang; Lei Li; Jing Zhang; Jun Li; Leaf Huang; et al. Nano-puerarin regulates tumor microenvironment and facilitates chemo- and immunotherapy in murine triple negative breast cancer model. Biomaterials 2020, 235, 119769-119769, 10.1016/j.biomaterials.2020.119769.
- Bo Han; Tao Wang; Zhigang Xue; Tao Wen; Ling Lu; Jie Meng; Jian Liu; Sizhu Wu; Jianchun Yu; Haiyan Xu; et al. Elemene Nanoemulsion Inhibits Metastasis of Breast Cancer by ROS Scavenging. International Journal of Nanomedicine 2021, ume 16, 6035-6048, 10.2147/ijn.s327094.
- Sofia M. Saraiva; Carlha Gutiérrez-Lovera; Jeannette Martínez-Val; Sainza Lores; Belén L. Bouzo; Sandra Díez-Villares; Sandra Alijas; Alba Pensado-López; Abi Judit Vázquez-Ríos; Laura Sánchez; et al. Edelfosine nanoemulsions inhibit tumor growth of triple negative breast cancer in zebrafish xenograft model. Scientific Reports 2021, 11, 1-13, 10.1038/s41598-021-87968-4.
- Supandeep Hallan; Maddalena Sguizzato; Elisabetta Esposito; Rita Cortesi; Challenges in the Physical Characterization of Lipid Nanoparticles. Pharmaceutics 2021, 13, 549, 10.3390/pharmaceutics13040549.
- Krishna Kumar Patel; Shilpkala Gade; Meraj Anjum; Sanjay Kumar Singh; Pralay Maiti; Ashish Kumar Agrawal; Effect of penetration enhancers and amorphization on transdermal permeation flux of raloxifene-encapsulated solid lipid nanoparticles: an ex vivo study on human skin. Applied Nanoscience 2019, 9, 1383-1394, 10.1007/s13204-019-01004-6.
- Shabi Parvez; Ganesh Yadagiri; Mallikarjuna Rao Gedda; Aakriti Singh; Om Prakash Singh; Anurag Verma; Shyam Sundar; Shyam Lal Mudavath; Modified solid lipid nanoparticles encapsulated with Amphotericin B and Paromomycin: an effective oral combination against experimental murine visceral leishmaniasis. Scientific Reports 2020, 10, 1-14, 10.1038/s41598-020-69276-5.
- Aakriti Singh; Ganesh Yadagiri; Shabi Parvez; Om Prakash Singh; Anurag Verma; Shyam Sundar; Shyam Lal Mudavath; Formulation, characterization and in vitro anti-leishmanial evaluation of amphotericin B loaded solid lipid nanoparticles coated with vitamin B12-stearic acid conjugate. Materials Science and Engineering: C 2020, 117, 111279, 10.1016/j.msec.2020.111279.
- Harshad Harde; Ashish Kumar Agrawal; Mahesh Katariya; Dnyaneshwar Kale; Sanyog Jain; Development of a topical adapalene-solid lipid nanoparticle loaded gel with enhanced efficacy and improved skin tolerability. RSC Advances 2015, 5, 43917-43929, 10.1039/c5ra06047h.
- Zehra Edis; Junli Wang; Muhammad Khurram Waqas; Muhammad Ijaz; Munazza Ijaz; Nanocarriers-Mediated Drug Delivery Systems for Anticancer Agents: An Overview and Perspectives. International Journal of Nanomedicine 2021, ume 16, 1313-1330, 10.2147/ijn.s289443.
- Zhe Cheng; Maoyu Li; Raja Dey; Yongheng Chen; Nanomaterials for cancer therapy: current progress and perspectives. Journal of Hematology & Oncology 2021, 14, 1-27, 10.1186/s13045-021-01096-0.
- Yongtao Duan; Abhishek Dhar; Chetan Patel; Mehul Khimani; Swarnali Neogi; Prolay Sharma; Nadavala Siva Kumar; Rohit L. Vekariya; A brief review on solid lipid nanoparticles: part and parcel of contemporary drug delivery systems. RSC Advances 2020, 10, 26777-26791, 10.1039/d0ra03491f.
- Ho Lun Wong; Reina Bendayan; Andrew M. Rauth; Yongqiang Li; Xiao Yu Wu; Chemotherapy with anticancer drugs encapsulated in solid lipid nanoparticles. Advanced Drug Delivery Reviews 2007, 59, 491-504, 10.1016/j.addr.2007.04.008.
- Anu Puri; Kristin Loomis; Brandon Smith; Jae-Ho Lee; Amichai Yavlovich; Eliahu Heldman; Robert Blumenthal; Lipid-Based Nanoparticles as Pharmaceutical Drug Carriers: From Concepts to Clinic. Critical Reviews in Therapeutic Drug Carrier Systems 2009, 26, 523-580, 10.1615/critrevtherdrugcarriersyst.v26.i6.10.
- Gamze Guney Eskiler; Gulsah Cecener; Unal Egeli; Berrin Tunca; Synthetically Lethal BMN 673 (Talazoparib) Loaded Solid Lipid Nanoparticles for BRCA1 Mutant Triple Negative Breast Cancer. Pharmaceutical Research 2018, 35, 218, 10.1007/s11095-018-2502-6.
- Venkata Talluri Siddhartha; Sai Kiran S. S. Pindiprolu; Pavan Kumar Chintamaneni; Shashank Tummala; S. Nandha Kumar; RAGE receptor targeted bioconjuguate lipid nanoparticles of diallyl disulfide for improved apoptotic activity in triple negative breast cancer: in vitro studies. Artificial Cells, Nanomedicine, and Biotechnology 2017, 46, 387-397, 10.1080/21691401.2017.1313267.
- Ishit R Kothari; Samrat Mazumdar; Saurabh Sharma; Kishan Italiya; Anupama Mittal; Deepak Chitkara; Docetaxel and alpha-lipoic acid co-loaded nanoparticles for cancer therapy. Therapeutic Delivery 2019, 10, 227-240, 10.4155/tde-2018-0074.
- Sai Kiran S. S. Pindiprolu; Pavan Kumar Chintamaneni; Praveen T. Krishnamurthy; Kinnera Ratna Sree Ganapathineedi; Formulation-optimization of solid lipid nanocarrier system of STAT3 inhibitor to improve its activity in triple negative breast cancer cells. Drug Development and Industrial Pharmacy 2018, 45, 304-313, 10.1080/03639045.2018.1539496.
- Sai Kiran Ss Pindiprolu; Praveen T Krishnamurthy; Venkata Rao Ghanta; Pavan Kumar Chintamaneni; Phenyl boronic acid-modified lipid nanocarriers of niclosamide for targeting triple-negative breast cancer. Nanomedicine 2020, 15, 1551-1565, 10.2217/nnm-2020-0003.
- Shilpkala Gade; Krishna Kumar Patel; Chandan Gupta; Meraj Anjum; Deepika Deepika; Ashish Kumar Agrawal; Sanjay Singh; An Ex Vivo Evaluation of Moxifloxacin Nanostructured Lipid Carrier Enriched In Situ Gel for Transcorneal Permeation on Goat Cornea. Journal of Pharmaceutical Sciences 2019, 108, 2905-2916, 10.1016/j.xphs.2019.04.005.
- Irma Danielle Rodrigues Pedro; Osmar Patricio Almeida; Helen Rodrigues Martins; Janaína De Alcântara Lemos; Andre Luis de Barros; Elaine Amaral Leite; Guilherme Carneiro; Optimization and in vitro/in vivo performance of paclitaxel-loaded nanostructured lipid carriers for breast cancer treatment. Journal of Drug Delivery Science and Technology 2019, 54, 101370, 10.1016/j.jddst.2019.101370.
- Qibo Zhang; Jihui Zhao; Hongmei Hu; Yulu Yan; Xiaoge Hu; Kuan Zhou; Sirui Xiao; Yong Tai Zhang; Nianping Feng; Construction and in vitro and in vivo evaluation of folic acid-modified nanostructured lipid carriers loaded with paclitaxel and chlorin e6. International Journal of Pharmaceutics 2019, 569, 118595, 10.1016/j.ijpharm.2019.118595.
- Eduardo Burgarelli Lages; Renata Salgado Fernandes; Juliana De Oliveira Silva; Ângelo Malachias de Souza; Geovanni Dantas Cassali; André Luís Branco de Barros; Lucas Antônio Miranda Ferreira; Co-delivery of doxorubicin, docosahexaenoic acid, and α-tocopherol succinate by nanostructured lipid carriers has a synergistic effect to enhance antitumor activity and reduce toxicity. Biomedicine & Pharmacotherapy 2020, 132, 110876, 10.1016/j.biopha.2020.110876.
- Shivaprasad Gadag; Reema Narayan; Archana S. Nayak; Diana Catalina Ardila; Shilpa Sant; Yogendra Nayak; Sanjay Garg; Usha Y. Nayak; Development and preclinical evaluation of microneedle-assisted resveratrol loaded nanostructured lipid carriers for localized delivery to breast cancer therapy. International Journal of Pharmaceutics 2021, 606, 120877, 10.1016/j.ijpharm.2021.120877.
- Sadaf Gilani; May Bin-Jumah; Rizwanullah; Syed Imam; Khalid Imtiyaz; Sultan Alshehri; Mohd. Rizvi; Chitosan Coated Luteolin Nanostructured Lipid Carriers: Optimization, In Vitro-Ex Vivo Assessments and Cytotoxicity Study in Breast Cancer Cells. Coatings 2021, 11, 158, 10.3390/coatings11020158.
- Sanyog Jain; Sandeep Kumar; Ashish Kumar Agrawal; Kaushik Thanki; Uttam Chand Banerjee; Enhanced Transfection Efficiency and Reduced Cytotoxicity of Novel Lipid–Polymer Hybrid Nanoplexes. Molecular Pharmaceutics 2013, 10, 2416-2425, 10.1021/mp400036w.
- Francesca Persano; Giuseppe Gigli; Stefano Leporatti; Lipid-polymer hybrid nanoparticles in cancer therapy: current overview and future directions. Nano Express 2021, 2, 012006, 10.1088/2632-959x/abeb4b.
- Tian Zhang; Preethy Prasad; Ping Cai; Chunsheng He; Dan Shan; Andrew Michael Rauth; Xiao Yu Wu; Dual-targeted hybrid nanoparticles of synergistic drugs for treating lung metastases of triple negative breast cancer in mice. Acta Pharmacologica Sinica 2017, 38, 835-847, 10.1038/aps.2016.166.
- Zilan Zhou; Carly Kennell; Joo-Youp Lee; Yuet-Kin Leung; Pheruza Tarapore; Calcium phosphate-polymer hybrid nanoparticles for enhanced triple negative breast cancer treatment via co-delivery of paclitaxel and miR-221/222 inhibitors. Nanomedicine: Nanotechnology, Biology and Medicine 2016, 13, 403-410, 10.1016/j.nano.2016.07.016.
- Neeraj K. Garg; Rajeev K. Tyagi; GajananD Sharma; Ashay Jain; Bhupinder Singh; Sanyog Jain; O. P. Katare; Functionalized Lipid–Polymer Hybrid Nanoparticles Mediated Codelivery of Methotrexate and Aceclofenac: A Synergistic Effect in Breast Cancer with Improved Pharmacokinetics Attributes. Molecular Pharmaceutics 2017, 14, 1883-1897, 10.1021/acs.molpharmaceut.6b01148.
- Filiz Bakar-Ates; Erva Ozkan; Ceyda Tuba Sengel-Turk; Encapsulation of cucurbitacin B into lipid polymer hybrid nanocarriers induced apoptosis of MDAMB231 cells through PARP cleavage. International Journal of Pharmaceutics 2020, 586, 119565, 10.1016/j.ijpharm.2020.119565.
This entry is offline, you can click here to edit this entry!