Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, General & Internal
|
Immunology
Conventional therapies for immune-mediated diseases, including autoimmune disorders, transplant reactions, and allergies, have undergone a radical evolution in the last few decades; however, they are still not specific enough to avoid widespread immunosuppression. The idea that vaccine usage could be extended beyond its traditional immunogenic function by encompassing the ability of vaccines to induce antigen-specific tolerance may revolutionize preventive and therapeutic strategies in several clinical fields that deal with immune-mediated disorders.
- tolerance
- immune response
- vaccine
1. Deprivation of Co-Stimulatory Signals
The most likely straightforward approach to induce peripheral tolerance is to deprive immune cells of co-stimulatory signals, as it allows them to become anergic and/or it promotes their conversion into Tregs. This has been achieved with the administration of artificial synthetic APCs, such as nanoparticles (NPs) (see below for a more detailed discussion about nanoparticles in tolerogenic vaccines), which exhibit antigens but lack costimulatory molecules on their surface [23,24]. In this sense, the immune response is not only dampened, but as this downregulation is antigen-specific, it ensures that immune anergy will only take place when that specific antigen is encountered [25] (Figure 1A).
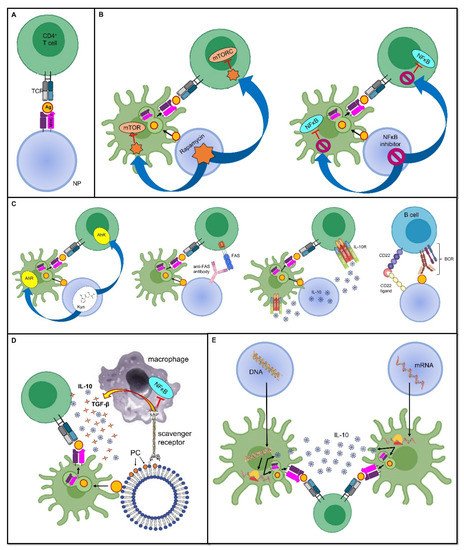
Figure 1. Tolerance-inducing strategies in tolerogenic vaccines. Mechanisms through which tolerogenic vaccines can induce an antigen-specific tolerogenic phenotype during the immune response. For the sake of simplicity, all vaccines are represented as being delivered through nanoparticles, even though several other delivery modes can be employed. Dimensions are not to scale. First, the antigen is usually endocytosed and processed by DCs; thereafter, the antigen is presented on MHC II and naive T cells. The tolerogenic adjuvant is delivered together with the antigen, and it has an effect on both the DCs and T cells interacting with them. (A) Deprivation of co-stimulatory signals. If the antigen is directly displayed on the MHC II molecule that is being carried by the vaccine, and in the absence of DC mediation and without co-stimulators, the T cell becomes anergic despite antigen recognition. (B) Inhibition of pro-inflammatory stimuli. Delivering the inhibitors of transcription factors that promote inflammation and cell proliferation is an additional strategy that can dampen the T cell response. (C) Induction of a tolerogenic phenotype. Engagement between signaling pathways are able to downregulate T or B cell activation, or it can induce programmed cell death. (D) Mimicry of tolerogenic physiological mechanisms. These strategies harness the physiological “neat death” (apoptosis) of cells; for instance, by displaying the apoptotic marker, PS, thus preventing an inappropriate inflammatory reaction. (E) Nucleic acid-based vaccines. The nanoparticle-delivered DNA coding for the specific antigen, as well as for a tolerogenic molecule, is first incorporated in the DC nucleus, then transcribed into an mRNA molecule, and eventually translated into a protein antigen plus the tolerogenic molecule. The former is loaded on an MHC II and presented to naive T cells, whereas the latter acts both on the DC itself and on naive T cells to induce a tolerogenic phenotype. In mRNA-based vaccines, the mechanism is very similar, with the only difference being that the transcription step is skipped, and therefore, the mRNA molecule is directly translated due to the DC ribosomes. Abbreviations. Ag, antigen; BCR, B cell receptor; CD22, cluster of differentiation 22; IL-10, interleukin-10; mTORC, mammalian target of rapamycin complex; NFκB, nuclear factor-κB; NP, nanoparticle; PC, phosphatidylcholine; TCR, T cell receptor; TGF-β, transforming growth factor-β.
Some promising results have been obtained in preclinical animal models of type 1 diabetes (T1D), MS, and arthritis. For instance, iron oxide nanoparticles coated with antigen-MHC I complexes allowed the suppression of autoreactive CD8+ T cells and their acquisition of anergic phenotypes. Similar results were obtained by coating particles with antigen-MHC II conjugates, resulting in the induction of Treg and regulatory B cells. Importantly, the transfer of these cells from vaccinated mice to naïve prediabetic animals conferred protection conferred protection to the development of diabetes, even when they had already experienced the antigen, thus suggesting that tolerance can be transferred and can reverse ongoing responses [23,24].
2. Inhibition of Pro-Inflammatory Stimuli
As stated above, inflammation provides a bridge between innate and adaptive immune responses as it foreruns the activation of T and B cells, and shapes their subsequent differentiation by providing the signals required for the complete induction of cell-mediated and humoral immunity. In this sense, using the inhibitors of pro-inflammatory mediators as adjuvants, which are co-delivered with antigens, may prove effective at inducing a specific tolerance against the antigens themselves. In a mouse model of inflammatory arthritis, Capini and colleagues showed that an injection of liposomes loaded with lipophilic nuclear factor-κB (NFκB) inhibitors, such as curcumin or quercetin, suppressed effector T cell responses, induced antigen-specific Tregs, and reversed the clinical signs of antigen-induced arthritis [26] (Figure 1B).
Furthermore, a tolerogenic scenario could be achieved by preventing the activity of immune cells at the level of their metabolism and cell cycles. An example of this is the use of the natural molecule rapamycin, derived from Streptomyces hygroscopicus, which behaves as an allosteric inhibitor of the mammalian target of the rapamycin (mTOR) complex-1 pathway, which is involved in cell proliferation and differentiation [27]. Combining the adjuvant rapamycin with antigen-containing vehicles, such as polymer particles, can inhibit T cell proliferation and promote Treg expansion, which was demonstrated in an experimental model of MS [28] (Figure 1B). Notably, the generalized immunosuppressive effect that this drug would have if administered alone is now finely tuned to be antigen-specific without inappropriately spreading the tolerogenic effect.
3. Harnessing Tolerogenic Physiological Mechanisms
Antigen delivery via the oral route has been demonstrated to be tolerogenic, similarly to the physiological tolerogenic response that is already present in healthy subjects, insofar as exogenous innocuous antigens and intestinal flora colonize the gut [13]. In the intestinal lamina propria and mesenteric lymph nodes, CD103+ Tregs are primed by CD103+ DCs through the release of TGF-beta and the expression of RALD2and the expression of the enzyme retinaldehyde dehydrogenase type 2 (RALD2), which turns vitamin A into retinoic acid (RA), a potent immunomodulator [29,30].
Allergens are initially orally administered in small doses which are then gradually increased. This is a well-known desensitizing approach that has been widely used to treat IgE- and Th2-mediated food allergies in order to modify the threshold for allergic sensitivity. With oral food challenges (OFC), the immune system becomes “used to the allergen”, and no longer reacts to it, or the immune system’s reaction is at least dampened; this allows the subject to enter a period of sustained unresponsiveness [31]. Harnessing a tolerogenic response towards oral antigens is a strategy that has also been applied to autoimmune disorders. Nevertheless, although oral tolerance appeared to be very effective in preclinical models, the results from human trials are still being investigated, and deserve further study [32]. For example, daily oral administration of bovine myelin in patients with relapsing–remitting (RR) MS reduced the frequency of myelin basic protein (MBP)-specific T cells in a phase I trial [33]; however, it did not improve clinical manifestations of MS in a larger phase III trial [34], and there are also some safety concerns regarding hypersensitive reactions [35]. Moreover, this strategy often requires long-lasting and repeated treatments since naked peptides are rapidly cleared and they only produce transient effects. Consequently, adjuvants would be required to improve their stability, bioavailability, and half-life [32].
As will be detailed in the following sections, nanoparticle-delivered antigens or antigen-coding DNA offer new hope; this is because coupling with NPs offers the advantage of providing an adjuvant to boost a specific tolerogenic response. Collagen-induced arthritis (CIA) was prevented in mice by oral administration using polylactic-co-glycolic acid (PLGA) particles loaded with collagen II (CII) 14 days before immunization with CII [36], whereas oral chitosan nanoparticles containing DNA coding for coagulation factor VIII (FVIII) were effective at inducing a sustained FVIII activity in the absence of neutralizing anti-FVIII antibodies in hemophilic mice [37].
Furthermore, other physiological processes can be mimicked to induce tolerance. For instance, since apoptotic cells generally induce a tolerogenic response, allowing a so-called “neat death” and preventing an inappropriate reaction against dying self-cells, harnessing apoptosis has been proposed for use in a mimicking approach when developing tolerogenic vaccine platforms [38]. This could be achieved by coupling antigens to splenocytes treated with ethylene carbodiimide (ECDI); this induces apoptosis, thus forming apoptotic cell–antigen conjugates that are able to induce antigen-specific tolerance [38]. Alternatively, the high turnover of red blood cells (RBCs), or eryptosis, can be exploited to allow antigens to strongly bind to RBCs; they are then processed in a tolerogenic manner by splenic T cells [39,40]. Moreover, using surrogate APCs, such as liposomes, displaying phosphatidylserine (PS) on their surface allows them to deceive macrophage PS-specific scavenger receptors and induce a tolerogenic phenotype; this includes an increase in anti-inflammatory IL-10 and TGF-β production and reduced pro-inflammatory NFκB and TNF-α signaling (Figure 1D). This model has been exploited to prevent the formation of inhibitory anti-FVIII antibodies in FVIII-treated hemophilic mice [41]. Promising results have also been obtained in mouse models of T1D [42] and experimental acute encephalomyelitis (EAE), which is the animal model of MS [43].
4. Induction of a Tolerogenic Phenotype
In addition to the imitation of naturally occurring tolerogenic mechanisms, it is also possible to shape the immune microenvironment through the co-delivery of antigens and anti-inflammatory cytokines, such as IL-10, or by engaging tolerogenic receptors. For instance, the tryptophan metabolite Kyn, which is involved in the activation of the tolerogenic Kyn–aryl hydrocarbon receptor (AhR) axis, has been used as an adjuvant. This is coupled to a phage vaccine expressing glutamic acid decarboxylase-65 (GAD65), one of the main autoantigens in T1D, and it has proven to be effective in the prevention of T1D in mouse models [44]. Similar encouraging results were obtained using latex beads coupled with class I MHC molecules and an anti-Fas monoclonal antibody mediating programmed cell death in a murine model of alloskin transplantation [45] (Figure 1C). Moreover, Macauley and colleagues have managed to obtain FVIII-specific tolerance in hemophilia mouse models by vaccinating them with liposomes carrying both FVIII and ligands of CD22 that inhibit the signaling of the BCR, thus dampening the humoral immune response. This tolerogenic vaccine prevented the formation of inhibitory antibodies to FVIII [46]. Of note, this approach is also compatible with the use of protein antigens because the CD22-mediated inhibition is sufficient to overwhelm their intrinsic immunogenicity [25] (Figure 1C). In general, the advantage of tolerance induction over the easier method of depriving the microenvironment of co-stimulatory signals is that, in this way, it is possible to force the immune microenvironment into a tolerogenic phenotype, even in the presence of strong pro-inflammatory stimuli [47].
5. Dendritic Cell-Based Vaccines
A key function in the complex interplay between innate and adaptive immunity is performed by APCs, whose prototype is represented by DCs. Indeed, DCs can be defined as a cellular bridge linking innate and adaptive immunity. On one hand, they can “sense the dangerous flavor” of either pathogens or cell damage by recognizing PAMPs and DAMPs through their PRRs. On the other hand, they possess the ability to process and display antigens that are loaded on the they possess the ability to process and diplay antigens to the TCR of T cells [48]. Upon stimulation, a maturation program is triggered, which includes the activation of the NF-κB or mTOR intracellular signaling pathways, culminating in the regulation of gene expression and the upregulation of all the necessary costimulators that are required for complete T cell activation (such as CD80 and CD86 and other costimulatory molecules) [27,49]. In addition, they can also determine the direction of the immune response by leaving either a pro-inflammatory or a tolerogenic footprint in the surrounding microenvironment through the release of a variety of cytokines [48].
Four major subtypes of DCs have been described: myeloid-derived type 1 and type 2 conventional DCs (cDC1s and cDC2s) are involved in the cross-presentation of antigens and CD8+ T cells, and the stimulation and polarization of CD4+ Th cells, respectively; lymphoid-derived plasmacytoid DCs (pDCs) quickly secrete type 1 interferons (IFN) in response to viral infections; and monocyte-derived DCs (moDCs) are differentiated from monocytes in the context of inflammation [2,50,51].
Although it was originally thought that immature (or resting) DCs induced T cell anergy via suboptimal antigen presentation and insufficient co-stimulation, the original dogma separating tolerogenic immature DCs from immunogenic mature and migratory DCs has been questioned [11]. This is because, in addition to a quantitative difference between MHC II and co-stimulatory molecule expression, a qualitative difference seems to be necessary for the induction of tolerance versus a pro-inflammatory response. Indeed, tolerogenic DCs possess a unique transcriptional program, resulting in a specific cytokine signature (TGF-β, IL-10), the release of immunosuppressive molecules such as nitric oxide, retinoic acid, and IDO, and the expression of ligands for inhibitory co-receptors (PD-L1/2, ICOSL, B7-H4, and B7-H3) that are able to induce the differentiation of T cells into Foxp3+ Treg [11,32,48]. Some authors have proposed that this qualitative subtype of DCs should be defined as “semi-mature” [52,53,54].
The most recently obtained knowledge concerning DC properties has paved the way for promising approaches in vaccine platform design. If DCs are key determinants in terms of initiating and mediating the adaptive immune response, it follows that they may be exploited and artificially modulated to serve either a tolerogenic or pro-inflammatory function, depending on what it required.
5.1. Ex Vivo DC Education
Ex vivo DC differentiation was one of the first attempts to design DC-based tolerogenic therapies. In brief, patient monocytes or their progenitors, which are recognizable from the expression of the hematopoietic cell marker, CD34, are cultured and allowed to be differentiated in a medium containing DC-dampening factors such as vitamin D, dexamethasone, or immunosuppressive cytokines such as IL-10 and TGF-β. Alternatively, they are genetically engineered to downregulate their expression of co-stimulatory molecules (CD80/86) [2,55]. If these growing DCs are also induced so that they encounter the disease-relevant antigen during the differentiation process, they become tolerogenic against a very specific target after re-infusion in the patient [56,57,58]. Similar attempts, which have been reviewed elsewhere [55], also included the expansion and subsequent reinfusion of mesenchymal stromal cells (MSCs) and Tregs. MSCs have a double advantage in that they lack both co-stimulatory molecules and MHC II expression, and they can be obtained from multiple sources, including from lipoaspirates. In allogeneic transplantation, MSCs obtained from the organ donor can be used to make the recipient more tolerant, thus counteracting the development of immune rejection [55,59].
So far, most studies have considered experimental models of autoimmune diseases in mice; although, some human clinical trials have been performed with promising results, mainly in the context of T1D [60], MS [61], Crohn’s disease [62], and rheumatoid arthritis [63,64,65]. Several aspects still need to be perfected, including optimal delivery route, whether to use parenteral or organ-targeting, the best timings for administration over the course of the disease, and the posology. In addition, the standardization of tolerogenic cell manufacturing techniques is still pending. In fact, despite good tolerability, ex vivo tolerogenic cell vaccination has some drawbacks, including the costly and cumbersome manufacturing process which requires controlled sterile conditions. This is coupled with the fact that monocyte-derived DCs are not exactly the same as their counterparts in vivo [2,66]. Moreover, in the field of Treg-based therapies, a possible safety concern involves the phenotypic instability of Tregs; this instability has been described in relation to the Tregs turning into pathogenic Th17 cells after repeated expansions, subsequently losing their tolerogenic potential, and exacerbating the disease [67,68].
5.2. In Vivo DC Targeting Strategies
An alternative to bypassing these obstacles is represented by in vivo DC vaccinations, which consist of DC targeting to induce them into acquiring a tolerogenic phenotype, but in an antigen-specific manner. This kind of DC “education” could be accomplished either through glycan–antigen or antibody–antigen conjugates or using nanoparticles as vectors for both the adjuvant and the antigen; this will be detailed in the following sections.
Among the PRRs of DCs, there is a subfamily of receptors bearing a carbohydrate recognition domain (CRD), including C-type lectin receptors (CLRs) and sialic acid-binding immunoglobulin-type lectins (Siglecs), which specifically recognize glycan moieties on host cells, pathogens, or allergens and they behave both as adhesion molecules and endocytic receptors. They may also mediate intracellular signaling pathways that can eventually instruct other immune cells. In particular, CLRs can engage with either immunoreceptor tyrosine-based activation motifs (ITAMs) or immunoreceptor tyrosine-base inhibitory motifs (ITIMs) depending on the ligand; these generate pro- or anti-inflammatory signals, respectively, whereas Siglecs predominantly produce anti-inflammatory signals using ITIMs or ITIM-like motifs [50].
Among some of the first studies on experimental models of autoimmune diseases, the utility of targeting the mannose receptor, DEC205, and mannose-, fucose-, and the n-acetylglucosamine-recognizing transmembrane protein, langerin (CD207), has been underlined. It seems that targeting these endocytic receptors on DCs using antigen–anti-DEC205 or antigen–anti-langerin conjugates promotes efficient uptake and presents antigens via the MHC I and MHC II pathways to CD8+ and CD4+ T cells, respectively. When applied to steady-state DCs, this approach leads to tolerance via different mechanisms, including dominant tolerance via the induction of Tregs, and passive tolerance via the induction of autoreactive T cell anergy and apoptosis [32,69,70]. Interestingly, langerin and DEC205 are often co-expressed, implying that it may be possible to exploit a dual targeting mechanism for Treg induction [71]. To list some examples, the injection of the EAE autoantigen, myelin oligodendrocyte glycoprotein (MOG), which was fused to anti-DEC205-specific antibodies, enhanced the antigen presentation via MHC II using steady-state DCs, which were induced to release IL-10 and TGF-β; this resulted in protection from induced EAE in 90% of the mice treated, compared with none in the control groups [72]. EAE symptom lessening was also obtained after targeting MOG with murine skin and lung DCs after conjugating it with anti-langerin antibodies; this always occurs via the induction of Foxp3+ Tregs [73]. Similar results were obtained in experimental models of inflammatory bowel disease (IBD) [74], autoimmune diabetes [75], and arthritis [76]. Unfortunately, data from human trials are yet to be obtained. The main obstacle derives from a differential and much broader expression pattern of DEC205 in human cells compared with murine cells [77,78,79], thus implying that there is a risk of offsite targeting.
DC carbohydrate receptor targeting can also be accomplished by using their natural carbohydrate ligands and coupling them with antigens. This is the case of DC-SIGN and mannose receptors (MRs), which are members of the CLR family that recognize mannose and fucose on many antigens. In particular, DC-SIGN is found only on immature DCs, and its targeting through fucosylated ligands prompts a Th2-biased anti-inflammatory response, Treg expansion, and inhibition of Th1/Th17 immunity [80,81].
Conversely, MR expression has been described on murine moDCs, macrophages, and CD1a+ dermal DC, and it seems capable of inducing an anti-inflammatory response through IL-10 secretion and PD-L1-mediated apoptosis of autoreactive T cells; this skews the immune response towards an increased Treg/Th1 ratio. Indeed, treatment with the soluble mannosylated proteolipid protein, M-PLP139–151, was shown to reduce both the incidence and severity of MS in a rodent model [82]. Vaccination with an epitope of a Leishmania analog, however, derived from the receptors of an activated C kinase (LACK), inhibited joint inflammation in an experimental model of autoimmune arthritis [83]. Similarly, encouraging results have been obtained in allergic disease settings using allergoid-mannan conjugates [47,84]; these results included data from clinical trials in humans (EudraCT numbers 2014-005471-88, 2015-000820-27, 2018-002522-23, and 2020-004126-32).
There is also compelling evidence that allergoid–mannan conjugates could drive the differentiation process of monocytes into tolerogenic or immunogenic moDCs through metabolic reprogramming and epigenetic modulation. A Spanish research group has recently explained that monocyte differentiation from nonatopic and allergic subjects, in the presence of grass pollen–mannan conjugates, yields tolerogenic moDCs with a higher expression of RNA in the typical tolerogenic molecules (IDO, PD-L1, IL-10). This occurs even after lipopolysaccharide (LPS) stimulation, together with a typical metabolic profile characterized by a decreased production of lactate, increased mitochondrial mass, and thus, a shift toward oxidative phosphorylation with greater ATP production, both before and after LPS-mediated stimulation. Moreover, chromatin immunoprecipitation (ChIP) analysis has allowed the molecular basis of epigenetic reprogramming to be simplified, as it relies mainly on histone modification, especially for IL-10 and PD-L1 enhancement, or miRNA involvement, particularly for TNF-α downregulation [47].
Among the ITIM-bearing DC receptors, some Siglecs could be triggered as a mechanism to downregulate the immune response in immune-mediated diseases. The broad Siglec family includes a variety of sialic acid-binding receptors that are differentially expressed on the many DC subtypes, and they show a differential affinity for sialic acid on its position on the underlying glycan (e.g., a better affinity with the α2,3 or α2,6 linkages), and it can perform either trans-interactions with sialic acid on different cells, or cis-interactions with ligands displayed on the same cell. Of note, these properties are particularly important to sustain paracrine and autocrine tolerogenic signaling, especially in steady-state DCs and Tregs, which are highly α2,6-sialylated. Through the ITIM-mediated engagement of the SH1- and SH2 domain-containing tyrosine phosphatases, (SHP1 and SHP2), Siglecs can boost the tolerance-inducing intracellular signaling pathways, which eventually leads to Treg induction and the decrease of pro-inflammatory Th1 and Th17 cell differentiation. Indeed, targeting pDCs via MOG–anti-Siglec H conjugates or sialylated MOG peptides resulted in dampened inflammatory responses in EAE mice [85,86], whereas the subcutaneous inoculation of sialic acid-modified grass pollen proved to be effective in terms of reducing allergic asthma in mice by reducing antigen-specific Th2 responses and eosinophilic accumulation in the airways [87]. Moreover, in this case, most results were confined to animal studies, and considerable effort should be made to translate these promising data to human settings. Notably, the associations between inhibitory signaling motifs allows Siglecs to behave as tolerogenic receptors even in a pro-inflammatory environment [86]. This is in contrast to other DC carbohydrate receptors such as DEC205, DC-SIGN, or langerin, which are tolerogenic only under steady-state conditions. Consequently, the appropriate context and formulation must be considered when designing tolerogenic vaccines.
6. Nucleic Acid-Based Tolerogenic Vaccines
An increased degree of complexity is achieved with nucleic acid-based vaccines that consist of DNA or mRNA molecules which encode the desired antigen(s), either alone or combined with immunomodulators. DNA- and RNA-based vaccines are first internalized by local or target cells. Subsequently, they use the cell machinery to translate into protein products, which are eventually post-translationally modified and subjected to traditional antigen presentation [32]. The expression of the gene of interest is controlled and promoted by coupling the coding sequence with a highly active promoter that is usually derived from cytomegalovirus (CMV). Moreover, with mRNA-based vaccines, antigen expression can be further enhanced by manipulating the mRNA molecule to include additional replicative sequences, most commonly from positive-stranded mRNA viruses, such as alphaviruses, which mediate mRNA auto-amplification and not only simple translation, thus obtaining a self-amplifying (SAM) mRNA vaccine [88].
Nucleic acid-based vaccines can either be administered naked, or packaged into microparticles or liposomes. This latter approach seems to improve their uptake and direct them toward the target site. The anatomical location of antigen recognition is of paramount importance since it can influence tolerance. Indeed, tolerogenic nucleic acid-based vaccines are often introduced in immunologically quiescent sites, such as the muscle, or in sites where Treg responses can be easily induced, such as the skin and the liver [32]. Furthermore, an additional step that is part of the manufacturing process of nucleic acid-based vaccines is the removal of intrinsic immunostimulatory components in the nucleotide sequences. For instance, extracellular and double-stranded RNA are inherently pro-inflammatory; therefore, efforts are made to remove double-stranded RNA contaminants to abrogate Toll-like receptor (TLR)-7 activation [89]. Similarly, to prevent TLR-3, TLR-7, and TLR-8 stimulation, uridine is replaced with 1-methylpseudouridine [90,91]. Additionally, in DNA, the number of immunostimulatory CpG motifs is reduced in order to limit TLR-9 activation, whereas immunoinhibitory GpG motifs are increased [92].
Intramuscular vaccination of EAE mice with a DNA vaccine encoding a MOG induced MOG-specific Treg expansion reduced the synthesis of IFN-γ, IL-17, and IL-4 after re-stimulation with MOG, thus reducing the clinical and histopathological signs of EAE [93]. The success obtained from the EAE results led to some phase I and phase II clinical trials that tested a DNA-based tolerogenic vaccine encoding MBP, which was found to be safe and able to decrease the number of central nervous system lesions in patients with RR–MS, although, the number was not high enough to be of statistical significance [92].
In addition to the antigen, nucleic acid-based vaccines can be engineered to encode immunomodulatory molecules as well, including IL-10, TGF-β, or IL-4, as adjuvants to ensure a tolerogenic response [32]. As an example, Schif-Zuck and colleagues managed to suppress MBP-induced EAE in rats that received separate plasmids encoding MBP68–86 or IL-10 under a CMV promoter, either prophylactically or therapeutically [94] (Figure 1E).
Nucleic acid-based vaccines have shown great promise in the prevention and management of immune-mediated disorders, even though the difficult control of the dose, the expression of the kinetics, and the instability of the mRNA molecule have hampered the enthusiasm generated by these methods, as their safety and efficacy remain controversial [92,94,95]. Nevertheless, the recent introduction of mRNA vaccines to prevent and alleviate the severity of the SARS-CoV-2 infection, and their efficacy in combating the COVID-19 pandemic, has broadened the application of nucleic acid-based vaccines. Hopefully, this experience could represent a good starting point in terms of improving knowledge and increasing the prophylactic and therapeutic use of tolerogenic vaccinations that are based on nucleic acids.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics14091782
This entry is offline, you can click here to edit this entry!