Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Although pancreatic cancer (PC) was considered in the past an orphan cancer type due to its low incidence, it may become in the future one of the leading causes of cancer death. Pancreatic ductal adenocarcinoma (PDAC) is the most frequent type of PC, being a highly aggressive malignancy and having a 5-year survival rate of less than 10%.
- pancreatic cancer
- risk factors
- tumor microenvironment
- PI3K/AKT/mTOR
1. Introduction
Pancreatic cancer (PC) is the 14th most frequent form of malignancy and represents the seventh most common cause of cancer mortality in the world, having the highest incidence in Europe and North America [1][2]. Although in the past it was considered an “orphan” cancer type because of its low incidence [3], by 2050 it may become one of the leading causes of cancer death [4]. In 2012, 338,000 people were diagnosed with PC, making it the 11th most common cancer worldwide [5]. In the United States, PC represents the third leading cause of cancer-related death, following colon and lung cancer, with an increase in both incidence and mortality, and it is forecast to become the second most common cause of cancer mortality by 2030 [6]. In Australia, PC has the peak mortality among all main cancer types as the fourth most frequent cause of mortality [7].
The first pancreatic tumor was described in 1971 [8]. Pancreatic tumors can be divided into two groups: endocrine and exocrine tumors [9][10]. Multiple endocrine neoplasia type 1 (MEN1), von Hippel–Lindau disease (VHL), neurofibromatosis 1 (NF-1) (von Recklinghausen disease), and the tuberous sclerosis complex (TSC) are pancreatic endocrine tumors (PETs) [11]. Adenoma, cystadenoma, lipoma, fibroma, hemangioma, lymphangioma, and neuroma are benign non-endocrine pancreas tumors [9]. Moreover, scientific data revealed that the nervous system is involved in PC carcinogenesis [12], so pancreatic tumors can be neuroendocrine neoplasms [13]. Therefore, pancreatic neuroendocrine tumors or PNETs develop from the endocrine component of the pancreas [10]. Pancreatic ductal adenocarcinoma (PDAC) and acinar cell carcinoma are pancreatic exocrine neoplasms [10]. In addition, PDAC derives from pre-malignant precursor lesions, known as intraepithelial neoplasms (PanINs) [14], which is the standard nomenclature and diagnostic criteria for duct lesions classification [15]. Together with PanIN, intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms (MCNs) are considered neoplastic precursors of the human PC [16].
PDAC is the most common type of PC, accounting for more than 90% of these neoplasms [17]. It is a highly aggressive malignancy, having a 5-year survival rate of less than 10% despite treatment [17]. This type of cancer is relatively uncommon, with an incidence of 8–12 per 100,000 per year and a lifetime risk of less than 3% of developing the disease; hence, the screening of asymptomatic adults is unfeasible [18]. Gastro-enteropancreatic neuroendocrine neoplasms (GEP-NENs) derive from neuroendocrine cells of the gastrointestinal tract [19]. Most of the PC cases arise from exocrine cells, while PC–endocrine cases including neuroendocrine tumors are uncommon [20].
The survival rate in PC remains low, with an improvement from <5% in 1990 to 9% in 2019 [18][21]. Currently, PC screening is not recommended for adult population because to its low incidence [22]. The only effective treatment of PC is the surgical resection of the tumor; therefore, an early diagnosis will improve the patient’s survival rate [23].
2. Tumor Microenvironment (TME) of Pancreatic Cancer
PC is a long, asymptomatic disease, so the development of initial carcinogenesis into invasive pancreatic carcinoma takes around 10 years. Therefore, in the absence of any screening method, this malignancy is discovered in late stages [24][25]. It has a high number of symptoms [26], with pain being frequent among PC patients due to pancreatic enzyme insufficiency, obstruction, and damage to the celiac plexus nerves [27]. Abdominal pain can have several causes, including tissue damage, inflammation, ductal obstruction, and infiltration [28]. Due to their abdominal localization, pancreatic tumors can be detected using computed tomography (CT), magnetic resonance imaging (MRI), or endoscopic ultrasound (EUS) even in early stages [24][29]. In the last decade, improvements have been achieved regarding PC chemotherapy, leading to a doubling in median overall survival rate [30].
The transformation of a normal cell into a malign one involves various mutations and epigenetic modifications [31]. The PC microenvironment contains fibroblasts, macrophages, and endothelial cells, increased numbers of extracellular matrix (ECM) compounds, elevated interstitial fluid pressure, and a high number of compressed tumor blood vessels [32]. Therefore, PDAC is characterized as a hard tumor [32]. In addition, pancreatic TME has an abundant fibrotic stroma, rich in various cell types and extracellular components including collagen, fibronectin, and hyaluronic acid [33]. Hyaluronan binds to its receptor CD44 and induces angiogenesis, epithelial-to-mesenchymal transition (EMT), and chemoresistance via receptor tyrosine kinase regulation and small GTPase [34].
Inflammation plays crucial roles in PC development from initiation to metastasis [35], leading to the activation of various inflammatory pathways [36]. Chronic inflammation induces the production of proinflammatory cytokines [37]. PDAC inflammation, which mediates TME development, involves exocrine acinar cells, ductal cells, and various stromal cells such as lymphocytes, neutrophils, mast cells, adipocytes, endothelial cells, and pancreatic stellate cells [38]. In a healthy state, stellate pancreatic cells are latent. In pancreatic pathological situations, stellate cells are capable of transitioning into a myofibroblast phenotype, playing a key role in the desmoplastic reaction [39]. PDAC stromal desmoplasia is the main histological hallmark, containing pancreatic stellate cells, ECM proteins, inflammatory cells, growth factors, and cytokines [40]. PDAC stromal desmoplastic reaction leads to the accumulation of a significant amount of type I collagen [41]. Moreover, via integrin α2β1, type I collagen is involved in PC cell adhesion, proliferation, and migration [34]. This cocktail of molecules and cells induces PDAC aggressiveness by promoting tumor growth, metastasis, and even chemoradiotherapy resistance [40].
In acute and chronic pancreatitis, acini, leukocytes, and stellate cells (Figure 1) contribute to pro-inflammatory cytokine release [42]. Therefore, PDAC chronic inflammation is caused by the risk factors mentioned above such as obesity, smoking, heavy drinking, low intake of fruits and vegetables, increased intake of fat, and a sedentary lifestyle [43]. The main mechanisms by which the microbiota influences PC development are chronic inflammation and antiapoptotic activity [44], regulation of the immune system-microbe-tumor axis, metabolism perturbation, and TME alteration [45].
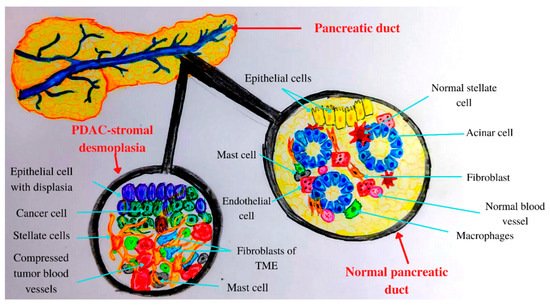
Figure 1. Anatomical and histological features of the pancreas: normal versus PADC.
Moreover, in the interstitial space of the tumor, PDAC patients have an excessive production of ECM proteoglycans and glycosaminoglycans [46]. ECM molecules that are secreted during tissue remodeling, angiogenesis, and tumor growth can modulate various cancer processes such as growth, metastasis, angiogenesis, differentiation, and immune response [47].
In PDAC, cancer-associated fibroblasts via various cytokines have an activated phenotype, promoting inflammation, angiogenesis, tumor growth and proliferation, invasion, metastasis, and regulation of the tumor metabolism [48]. The immune system is also involved, releasing cytokines, chemokines, and growth factors, which can mediate the neoplastic transformation [31]. Inflammation and insulin resistance, associated with obesity and type 2 diabetes, can enhance KRAS activation [49]. Moreover, a high fat diet induces KRAS activation, transforming a normal pancreatic cell into pancreatic intraepithelial neoplastic lesions [49]. Furthermore, TME orchestrates the tumor cell transcription programs [50]. EMT occurs in response to the activation of several pathways, such as receptor tyrosine pathways and TGF-β [51].
In the periphery of PDAC, tumor-associated macrophages are accumulated, being correlated with a M2 phenotype. This type of phenotype is characterized by extra-pancreatic and lymph vessel invasion, lymph node involvement, and shortened survival time [52]. The adipose tissue may also contribute to PC development via adipocytes, immune cells, endothelial cells, fibroblasts, stem cells, or progenitor cells [53]. Many of these cells can secrete pro-inflammatory cytokines (TNF-α, IL-6, leptin, TGF-β), which are known to be involved in cancer proliferation [53].
Exosomes, which are involved in the transport of proteins, lipids, and nucleic acids between cells, may be secreted by tumor cells including PC. PDAC tumor exosomes are involved in the establishment of the immunosuppressive microenvironment [54].
Among screening biomarkers, serum carbohydrate antigen 19-9 (CA 19-9) is the most used biomarker for PC, with a median sensitivity of 79% (70–90%) [55]. Kurahara H. and his research team detected CA 19-9 and p53 in 115 patients (80 with upfront surgery and 35 who received neoadjuvant therapy for resectable PC) [56]. These two biomarkers were statistically elevated in resectable PC patients with early recurrence after upfront surgery [56]. N6-methyladenosine (m6A) is the most recurrent RNA modification found in eukaryotes and is involved in the regulation of many cellular and biological processes and detected in various malignancies including PC [57].
3. PI3K/AKT/mTOR and Pancreatic Cancer
Cells and tissues communicate with each other by binding of various molecules—such as insulin, glucose, growth factors, cytokines [48], integrins, B and T cell receptors [58], hormones, and chemokines—to the tyrosine kinases (RTKs) or G-protein receptors (GPCRs) at the cell surface. This leads to the activation of intracellular signaling pathways, such as the class I phosphoinositide 3-kinase (PI3K)/Protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway [59][60][61].
PI3Ks are divided into three classes [62], each having four homologous regions, where the kinase domain is the most conserved [63]. In addition, PI3K class I has two subclasses, PI3K class IA that is activated by RTKs and PI3K class IB which is activated by GPCRs [64], and both are implicated in cancerous cell growth, survival, and invasion [65]. PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3), leading to AKT activation [66]. PTEN is a lipid phosphatase that acts on PIP3, causing its dephosphorylation, thus being considered the negative regulator of the signaling pathway [67].
AKT activation consists of two phosphorylation processes, one performed by mammalian target of rapamycin complex 2 (mTORC2) at serine 473 residue, and the second one at threonine 308 residue performed by phosphoinositide-dependent kinase 1 (PDK1) [68]. In eukaryotes, AKT kinase has three isoforms, AKT1, AKT2, and AKT3, with high homology [68]. The first two isoforms are ubiquitously expressed. AKT2 is predominantly found in insulin-responsive cells, while AKT3 seems to be found mostly in neurons and testes [69]. AKT1 and AKT2 gene mutations are implicated in insulin resistance and further in diabetes pathogenesis [70]. AKT activation induces tuberous sclerosis protein 2 (TSC2) inhibition [71] and disrupt its interaction with TSC1 [72]. TSC1 is involved in cell growth and proliferation, survival, and autophagy [73]. Once activated, AKT induces protein synthesis and cell growth by mTOR activation and TSC1 and TSC2 inhibition [74].
mTOR has two complexes: mTORC1 complex, composed of mTOR, raptor, mLST8, and PRAS40, and the second complex mTORC2, which is formed by mTOR, rictor, mLST8, and Sin1 [75]. mTOR regulates translation by the phosphorylation of ribosomal protein S6 kinases (S6K) and 4E-binding protein 1 (4E-BP1). Activation of 4E-BP1 induces the release of the eukaryotic translation initiation factor 4E (eIF4E) [76]. The mTOR pathway is the main regulator of the mammalian metabolism that regulates cell growth and proliferation, protein synthesis, and autophagy [77]. mTOR plays an important role in apoptosis regulation, stimulating the cell growth [65] and immune cell regulation, differentiation, and functions [78]. Growth factors, nutrient energy, and even stress may activate or inactivate mTOR, which induces normal or dysregulated processes [79].
In the pathogenesis of PC, several key signaling pathways are involved, such as PI3K/AKT/mTOR [80] and Notch, Wnt, and Hedgehog [81]. Overexpression of the PI3K/AKT/mTOR pathway has been observed in a large number of malignancies including PC [82][83][84][85][86][87][88]. In addition, PI3K/AKT/mTOR is one of the most mutated signaling pathways in human cancers, usually correlated with the loss of PTEN and mutations in PIK3CA (encoding PI3K-p110α) and AKT1 [89]. PTEN expression is lost in 25–70% of all PC cases [90]. Furthermore, KRAS mutation induces overexpression of mutant PI3KCA [65].
One of the numerous signaling pathways activated by KRAS mutation is PI3K, which is also stimulated by various cytokines and growth factors through the RTKs [65]. The overexpression of AKT1, which stimulates cancer cell growth and proliferation, was found in 10–20% of the patients with PDAC [65]. During tumorigenesis, AKT1 may have opposite effects, may act as a pro-oncogenic factor suppressing apoptosis, or may restrict the tumor invasion [91]. Regarding AKT2, this isoform promotes cell migration and invasion, while AKT3 is involved in tumor migration [91].
Therefore, the PI3K/AKT/mTOR signaling pathway plays crucial roles in cell survival, growth, proliferation [92], metabolism, and motility (Figure 2) [93]. Moreover, these protein kinases, through phosphorylation processes, are involved in various cellular functions such as transcription and translation [94]. AKT is involved in cell survival and apoptosis by regulating some pro-survival and anti-apoptotic proteins Bcl-XL and NF-kB in both normal and cancerous cells, so targeting this molecule is considered a promising therapeutic approach for the treatment of pancreatic ductal adenocarcinoma [95].
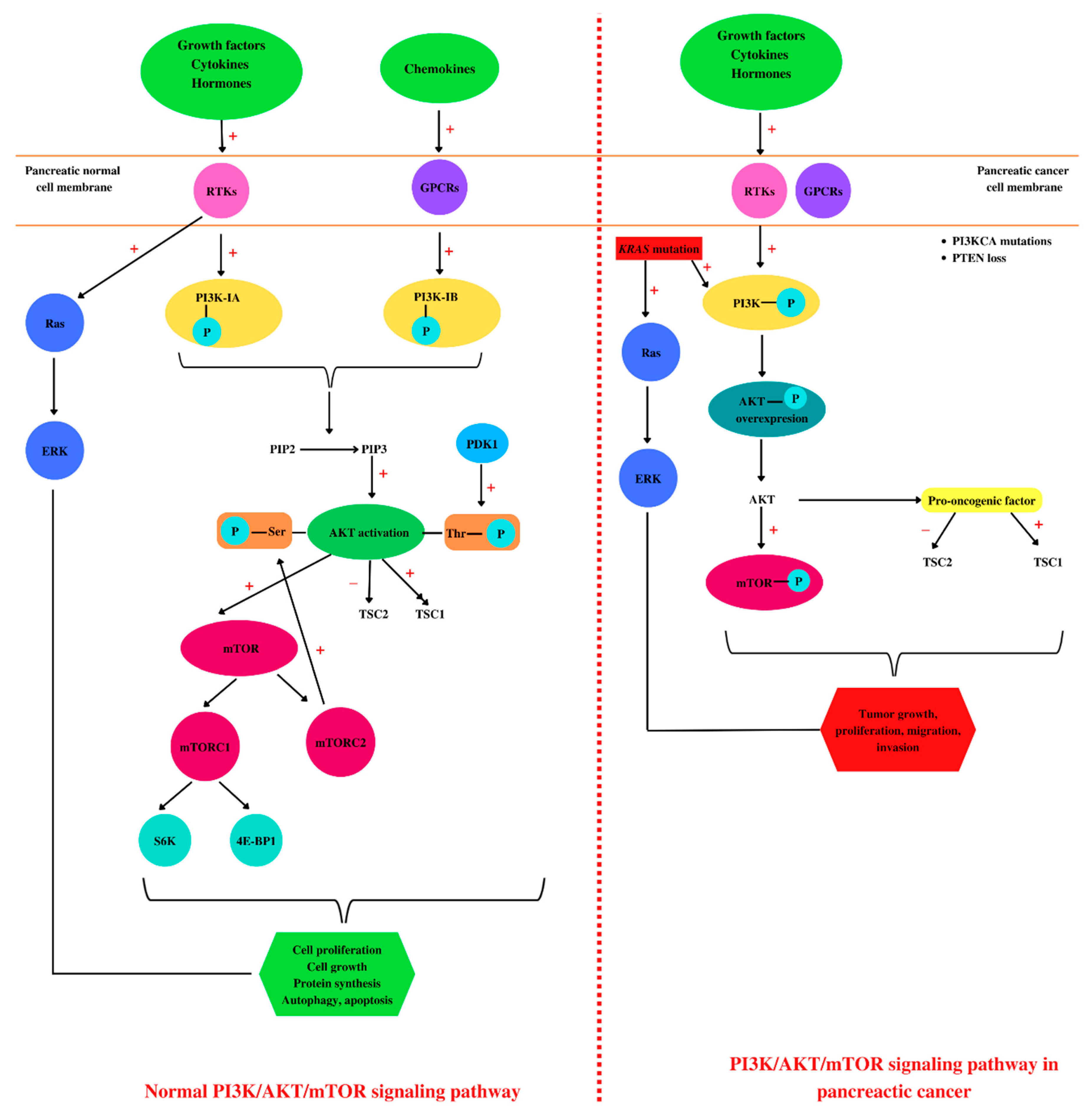
Figure 2. Pancreatic PI3K/AKT/mTOR: normal versus pathological. In a healthy cell, such as a pancreatic cell, different ligands (growth factors, hormones, cytokines, or chemokines) bind to tyrosine kinase (RTKs) or G-protein receptors (GPCRs) located in the cell membrane, leading to PI3K activation by phosphorylation. Further, PI3K catalyzes the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) at phosphatidylinositol 3,4,5-trisphosphate (PIP3), leading to AKT activation. AKT activation consists of two phosphorylation processes, one performed by mammalian target of rapamycin complex 2 (mTORC2) at serine 473 residue and the second one at threonine 308 residue performed by phosphoinositide-dependent kinase 1 (PDK1). Phosphorylated AKT induces further activation of mTOR and tuberous sclerosis protein 1 (TSC1) and TSC2 inhibition. In addition, mTOR phosphorylates ribosomal protein S6 kinases (S6K) and 4E-binding protein 1 (4E-BP1). The binding of growth factors to RTKs leads to Ras activation and in the end to extracellular signal-regulated kinase (ERK) activation, correlated with cell proliferation. Altogether, ERK and AKT activation are conducive to cell growth, proliferation, protein synthesis, autophagy, and apoptosis. In pancreatic cancer cells, besides growth factors, hormones, and cytokines, KRAS mutations activate PI3K and Ras. Moreover, these types of cells are characterized by PIK3CA mutations and PTEN loss. All these molecular events are associated with an overexpression of AKT phosphorylation, regarded as a pro-oncogenic factor. Together with ERK, AKT induces tumor growth, proliferation, migration, and invasion. “+”: activates, “−”: inhibits.
In pathological conditions, cytokines, chemokines, and Fc receptors activate the PI3K/AKT/mTOR signaling pathway [96]. Vascular endothelial growth factor (VEGF), IGF-1, FGF, platelet-derived growth factor (PDGF), and TGF-α and -β bind to RTKs and activate PI3Ks [62][97][98]. VEGF and PDGF induce angiogenesis in neuroendocrine tumors, contributing to neuroendocrine tumor development [99]. PI3K, AKT, and mTOR are the three major nodes that suffer dysregulation and induce cancer progression [100]. Moreover, genomic aberrations activate PI3K in various PC lineages [101] (Figure 3).
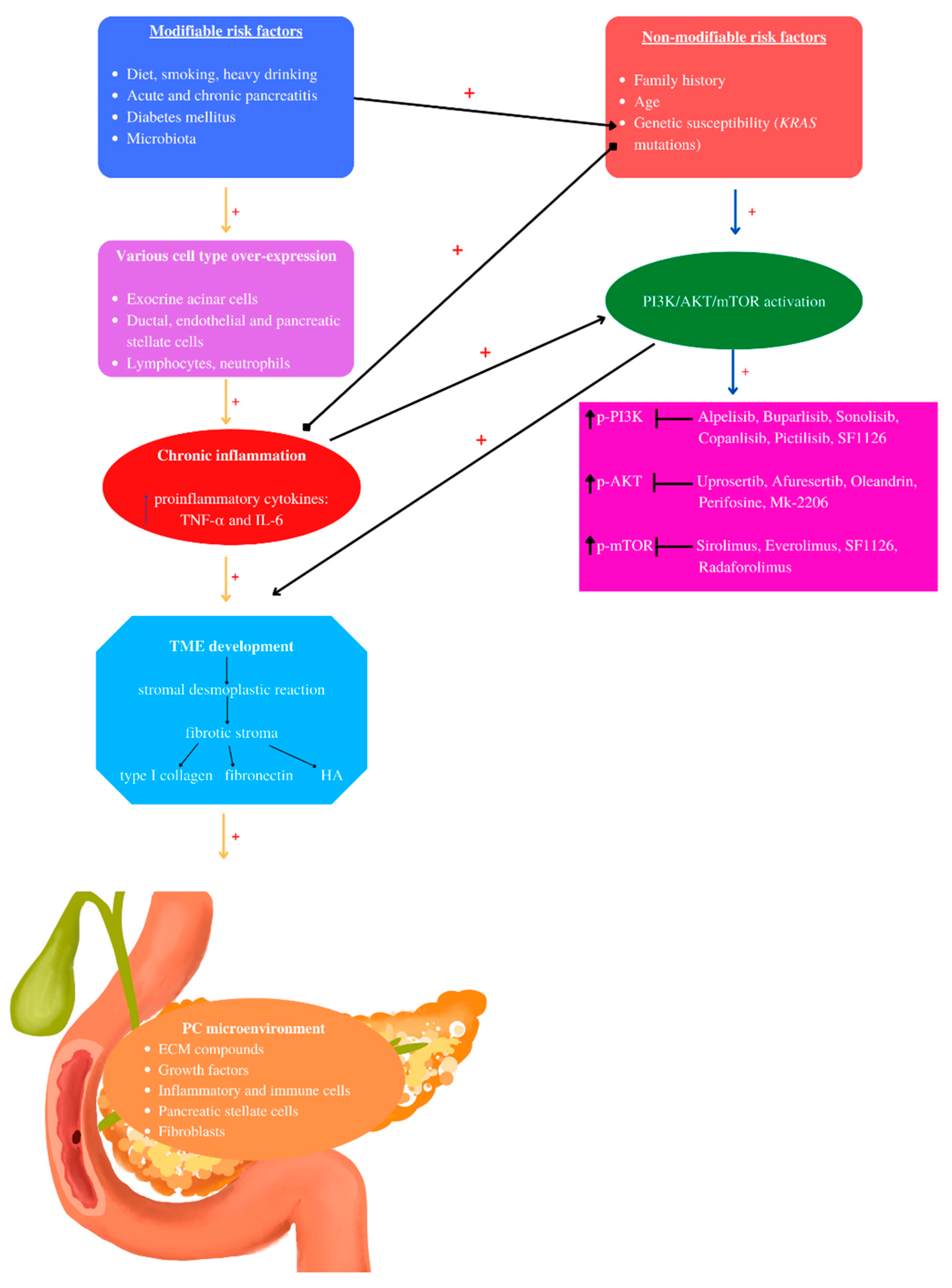
Figure 3. Pancreatic cancer pathogenesis: The pathogenesis of PC involves non- and modifiable risk factors that induce chronic inflammation mediated by several cell types. Chronic inflammation mediates TME development characterized by stromal desmoplastic reaction—the main PC histological hallmark, leading to fibrotic stoma formation, rich in type I collagen, fibronectin, and hyaluronic acid (HA). KRAS mutations may be activated by inflammation and modifiable risk factors such as diet and diabetes mellitus. Chronic inflammation and KRAS mutations are the main actors that induce PI3K/AKT/mTOR activation, contributing to PC-TME development, correlated with increased phosphorylated levels. Currently, the mentioned PI3K/AKT/mTOR inhibitors have been used in clinical studies. “+” activates.
Prorenin receptor (P)RR is a single transmembrane protein encoded by the ATP6AP2 gene located on the X chromosome [102]. (P)RR is widely expressed in the brain, pancreas, heart, liver, placenta, and kidney, with this expression being significantly higher in various human cancers. (P)RR may activate PI3K/AKT/mTOR via angiotensin-II-reactive oxygen species formation [102]. The aldehyde dehydrogenase 1 family member A3 (ALDH1A3) is a crucial enzyme for glucose metabolism that can increase the expression of hexokinase 2 and further enhance glycolysis in PDAC cells. During pancreatic pathogenesis, glucose metabolism may promote PDAC metastasis both in vitro and in vivo [103]. Therefore, Nie S. et al. detected that ALDH1A3 is overexpressed in vitro and leads to increased activity of PI3K/AKT/mTOR [103].
In the liver, increased insulin levels enhance insulin-like growth factor-1 (IGF-1) synthesis and down-regulate IGF-1 binding proteins, leading to cell proliferation and an increased risk of PC [104]. Yan X. and his research team evaluated the expression of EG-VEGF in PC tissues and cells. The study reported high expression of EG-VEGF in PDAC, both cells and tissues. Moreover, the inhibition of EG-VEGF inhibits PC cell proliferation and promotes apoptosis via PI3K/AKT/mTOR [105].
Su CC measured the levels of EGFR, IGF1R, VEGFR, PI3K, AKT, mTOR, and PTEN in MiaPaCa2 human PC cells and observed their increased expression [106]. Lin S. and co-workers obtained similar results regarding PI3K, p-AKT, and p-mTOR using human PC cell lines (PANC-1). Moreover, the study reported that PC cell line supplementation with IGF-1 significantly increases the expression of the three proteins mentioned above [107]. Poor PC prognosis is linked to AKT phosphorylation at S473 [108]. Singh BN and co-workers detected increased phosphorylated levels of AKT and mTOR in human PC stem cells [109]. Liu X. and his research team measured elevated activity of PI3K/Akt/mTOR pathway in tissue samples of 54 PDAC patients [110]. In addition, serine/threonine kinase 33 (STK33) is involved in pancreatic neuroendocrine tumor (PNET) growth and progression via PI3K/AKT/mTOR pathway activation. Thus, STK33 was overexpressed in PNET specimens compared with normal pancreatic tissue [111].
This entry is adapted from the peer-reviewed paper 10.3390/ijms231710132
References
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World. J. Gastroenterol. 2018, 21, 4846–4861.
- Furuse, J.; Nagashima, F. Emerging protein kinase inhibitors for treating pancreatic cancer. Expert. Opin. Emerg. Drugs. 2017, 22, 77–86.
- Lowenfels, A.B.; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best. Pract. Res. Clin. Gastroenterol. 2006, 20, 197–209.
- Korc, M.; Jeon, C.Y.; Edderkaoui, M.; Pandol, S.J.; Petrov, M.S. Tobacco and alcohol as risk factors for pancreatic cancer. Best. Pract. Res. Clin. Gastroenterol. 2017, 31, 529–536.
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World. J. Gastroenterol. 2016, 28, 9694–9705.
- Morrison, A.H.; Byrne, K.T.; Vonderheide, R.H. Immunotherapy and prevention of pancreatic cancer. Trends Cancer 2018, 4, 418–428.
- Loveday, B.P.T.; Lipton, L.; Thomson, B.N. Pancreatic cancer: An update on diagnosis and management. Aust. J. Gen. Pract. 2019, 48, 826–831.
- Torphy, R.J.; Fujiwara, Y.; Schulick, R.D. Pancreatic cancer treatment: Better, but a long way to go. Surg. Today 2020, 50, 1117–1125.
- Goral, V. Pancreatic cancer: Pathogenesis and Diagnosis. Asian. Pac. J. Cancer Prev. 2015, 16, 5619–5624.
- Lanfredini, S.; Thapa, A.; O’Neill, E. RAS in pancreatic cancer. Biochem. Soc. Trans. 2019, 30, 961–972.
- Jensen, R.T.; Berna, M.J.; Bingham, D.B.; Norton, J.A. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 2008, 1, 1807–1843.
- Wakiya, T.; Ishido, K.; Yoshizawa, T.; Kanda, T.; Hakamada, K. Roles of the nervous system in pancreatic cancer. Ann. Gastroenterol. Surg. 2021, 29, 623–633.
- Clift, A.K.; Kidd, M.; Bodei, L.; Toumpanakis, C.; Baum, R.P.; Oberg, K.; Modlin, I.M.; Frilling, A. neuroendocrine neoplasms of the small bowel and pancreas. Neuroendocrinology 2020, 110, 444–476.
- Kennedy, A.L.; Adams, P.D.; Morton, J.P. Ras, PI3K and senescence. Small GTPase 2011, 2, 264–267.
- Zavoral, M.; Minarikova, P.; Zavada, F.; Salek, C.; Minarik, M. Molecular biology of pancreatic cancer. World. J. Gastroenterol. 2011, 28, 2897–2908.
- Ariston, G.A.N.; Jiao, Q.; Yvette, U.; Yang, X.; Al-Ameri, S.A.; Du, L.; Wang, C. Differences between KC and KPC pancreatic ductal adenocarcinoma mice models, in terms of their modeling biology and their clinical relevance. Pancreatology 2020, 20, 79–88.
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World. J. Gastrointest. Oncol. 2020, 15, 173–181.
- Pereira, S.P.; Oldfield, L.; Ney, A.; Hart, P.A.; Keane, M.G.; Pandol, S.J.; Li, D.; Greenhalf, W.; Jeon, C.Y.; Koay, E.J.; et al. Early detection of pancreatic cancer. Lancet Gastroenterol. Hepatol. 2020, 5, 698–710.
- Zanini, S.; Renzi, S.; Giovinazzo, F.; Bermano, G. mTOR pathway in gastroenteropancreatic neuroendocrine numor (GEP-NETs). Front. Endocrinol. 2020, 16, 562505.
- Galli, R.; Schmutz-Kober, K.; Kühnel, J.; Maak, M.; Rosenberg, R. Pancreatic cancer. Ther. Umsch. 2021, 78, 605–613.
- Biller, L.H.; Wolpin, B.M.; Goggins, M. Inherited pancreatic cancer syndromes and high-risk screening. Surg. Oncol. Clin. N. Am. 2021, 30, 773–786.
- Garg, S.K.; Chari, S.T. Early detection of pancreatic cancer. Curr. Opin. Gastroenterol. 2020, 36, 456–461.
- Cai, J.; Chen, H.; Lu, M.; Zhang, Y.; Lu, B.; You, L.; Zhang, T.; Dai, M.; Zhao, Y. Advances in the epidemiology of pancreatic cancer: Trends, risk factors, screening, and prognosis. Cancer Lett. 2021, 1, 1–11.
- Lang, J.; Kunovský, L.; Kala, Z.; Trna, J. Risk factors of pancreatic cancer and their possible uses in diagnostics. Neoplasma 2021, 68, 227–239.
- Lee, E.S.; Lee, J.M. Imaging diagnosis of pancreatic cancer: A state-of-the-art review. World. J. Gastroenterol. 2014, 28, 7864–7877.
- Carvajal, G. Pancreatic cancer related pain: Review of pathophysiology and intrathecal drug delivery systems for pain management. Pain Physician 2021, 24, E583–E594.
- Coveler, A.L.; Mizrahi, J.; Eastman, B.; Apisarnthanarax, S.J.; Dalal, S.; McNearney, T.; Pant, S.; Precision Promise Consortium. Pancreas cancer-associated pain management. Oncologist 2021, 26, e971–e982.
- Lohse, I.; Brothers, S.P. Pathogenesis and treatment of pancreatic cancer related pain. Anticancer. Res. 2020, 40, 1789–1796.
- Moore, A.; Donahue, T. Pancreatic Cancer. JAMA 2019, 8, 322, 1426.
- Gupta, R.; Amanam, I.; Chung, V. Current and future therapies for advanced pancreatic cancer. J. Surg. Oncol. 2017, 116, 25–34.
- Kaur, S.; Kumar, S.; Momi, N.; Sasson, A.R.; Batra, S.K. Mucins in pancreatic cancer and its microenvironment. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 607–620.
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental determinants of pancreatic cancer. Physiol. Rev. 2020, 1, 1707–1751.
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348.
- Xu, Z.; Pothula, S.P.; Wilson, J.S.; Apte, M.V. Pancreatic cancer and its stroma: A conspiracy theory. World. J. Gastroenterol. 2014, 28, 11216–112129.
- Shi, J.; Xue, J. Inflammation and development of pancreatic ductal adenocarcinoma. Chin. Clin. Oncol. 2019, 8, 19.
- Tamtaji, O.R.; Mirhosseini, N.; Reiter, R.J.; Behnamfar, M.; Asemi, Z. Melatonin and pancreatic cancer: Current knowledge and future perspectives. J. Cell. Physiol. 2019, 234, 5372–5378.
- Orlacchio, A.; Mazzone, P. The Role of Toll-like Receptors (TLRs) mediated inflammation in pancreatic cancer pathophysiology. Int. J. Mol. Sci. 2021, 25, 12743.
- Tao, X.; Xiang, H.; Pan, Y.; Shang, D.; Guo, J.; Gao, G.; Xiao, G.G. Pancreatitis initiated pancreatic ductal adenocarcinoma: Pathophysiology explaining clinical evidence. Pharmacol. Res. 2021, 168, 105595.
- Ferdek, P.E.; Jakubowska, M.A. Biology of pancreatic stellate cells-more than just pancreatic cancer. Pflugers Arch. 2017, 469, 1039–1050.
- Khetan, K.; Baloda, V.; Sahoo, R.K.; Vishnubhathla, S.; Yadav, R.; Saraya, A.; Sharma, A.; Gupta, S.D.; Das, P. SPARC expression in desmoplastic and non desmoplastic pancreatic carcinoma and cholangiocarcinoma. Pathol. Res. Pract. 2019, 215, 152685.
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA + myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 12, 548–565.e6.
- Murtaugh, L.C.; Keefe, M.D. Regeneration and repair of the exocrine pancreas. Ann. Rev. Physiol. 2015, 77, 229–249.
- Li, J.; Chen, X.; Kang, R.; Zeh, H.; Klionsky, D.J.; Tang, D. Regulation and function of autophagy in pancreatic cancer. Autophagy 2021, 17, 3275–3296.
- Karpiński, T.M. The microbiota and pancreatic cancer. Gastroenterol. Clin. North. Am. 2019, 48, 447–464.
- Wei, M.Y.; Shi, S.; Liang, C.; Meng, Q.C.; Hua, J.; Zhang, Y.Y.; Liu, J.; Zhang, B.; Xu, J.; Yu, X.J. The microbiota and microbiome in pancreatic cancer: More influential than expected. Mol. Cancer 2019, 20, 97.
- Suklabaidya, S.; Dash, P.; Das, B.; Suresh, V.; Sasmal, P.K.; Senapati, S. Experimental models of pancreatic cancer desmoplasia. Lab. Investig. 2018, 98, 27–40.
- Kaps, L.; Schuppan, D. Targeting cancer associated fibroblasts in liver fibrosis and liver cancer using nanocarriers. Cells 2020, 3, 2027.
- Sun, K.; Luo, J.; Guo, J.; Yao, X.; Jing, X.; Guo, F. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A narrative review. Osteoarthr. Cartil. 2020, 28, 400–409.
- Dehghanian, F.; Azhir, Z.; Khalilian, S.; Grüning, B. Non-coding RNAs underlying the pathophysiological links between type 2 diabetes and pancreatic cancer: A systematic review. J. Diabetes. Investig. 2022, 13, 405–428.
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, 102655.
- Krantz, S.B.; Shields, M.A.; Dangi-Garimella, S.; Munshi, H.G.; Bentrem, D.J. Contribution of epithelial-to-mesenchymal transition and cancer stem cells to pancreatic cancer progression. J. Surg. Res. 2012, 173, 105–112.
- Ma, X.; Wu, D.; Zhou, S.; Wan, F.; Liu, H.; Xu, X.; Xu, X.; Zhao, Y.; Tang, M. The pancreatic cancer secreted REG4 promotes macrophage polarization to M2 through EGFR/AKT/CREB pathway. Oncol. Rep. 2016, 35, 189–196.
- Xu, M.; Jung, X.; Hines, O.J.; Eibl, G.; Chen, Y. Obesity and pancreatic cancer: Overview of epidemiology and potential prevention by weight loss. Pancreas 2018, 47, 158–162.
- Batista, I.A.; Melo, S.A. Exosomes and the future of immunotherapy in pancreatic cancer. Int. J. Mol. Sci. 2019, 29, 567.
- Chu, D.; Kohlmann, W.; Adler, D.G. Identification and screening of individuals at increased risk for pancreatic cancer with emphasis on known environmental and genetic factors and hereditary syndromes. JOP 2010, 5, 203–212.
- Kurahara, H.; Maemura, K.; Mataki, Y.; Sakoda, M.; Iino, S.; Kawasaki, Y.; Arigami, T.; Mori, S.; Kijima, Y.; Ueno, S.; et al. A therapeutic strategy for resectable pancreatic cancer based on risk factors of early recurrence. Pancreas 2018, 47, 753–758.
- Li, J.; Wang, F.; Liu, Y.; Wang, H.; Ni, B. N6-methyladenosine (m6A) in pancreatic cancer: Regulatory mechanisms and future direction. Int. J. Biol. Sci. 2021, 4, 2323–2335.
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907.
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555.
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The relation between PI3K/AKT signalling pathway and cancer. Gene 2019, 25, 120–128.
- Sharma, A.; Mehan, S. Targeting PI3K-AKT/mTOR signaling in the prevention of autism. Neurochem. Int. 2021, 147, 105067.
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 6, 1483–1496.
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791.
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518.
- Mehra, S.; Deshpande, N.; Nagathihalli, N. Targeting PI3K pathway in pancreatic ductal adenocarcinoma: Rationale and progress. Cancers 2021, 2, 4434.
- Hung, S.W.; Zhang, R.; Tan, Z.; Chung, J.P.W.; Zhang, T.; Wang, C.C. Pharmaceuticals targeting signaling pathways of endometriosis as potential new medical treatment: A review. Med. Res. Rev. 2021, 41, 2489–2564.
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug. Targets 2008, 8, 187–198.
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell Physiol. 2019, 234, 2373–2385.
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal 2019, 21, 154.
- Peng, Z.; Aggarwal, R.; Zeng, N.; He, L.; Stiles, E.X.; Debebe, A.; Chen, J.; Chen, C.Y.; Stiles, B.L. AKT1 regulates endoplasmic reticulum stress and mediates the adaptive response of pancreatic β Cells. Mol. Cell. Biol. 2020, 14, e00031-20.
- De Santis, M.C.; Sala, V.; Martini, M.; Ferrero, G.B.; Hirsch, E. PI3K Signaling in Tissue hyper-proliferation: From overgrowth syndromes to kidney cysts. Cancers 2017, 29, 30.
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657.
- Mallela, K.; Kumar, A. Role of TSC1 in physiology and diseases. Mol. Cell. Biochem. 2021, 476, 2269–2282.
- Makker, A.; Goel, M.M.; Mahdi, A.A. PI3K/PTEN/Akt and TSC/mTOR signaling pathways, ovarian dysfunction, and infertility: An update. J. Mol. Endocrinol. 2014, 53, R103–R118.
- Owonikoko, T.K.; Khuri, F.R. Targeting the PI3K/AKT/mTOR pathway: Biomarkers of success and tribulation. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, e395–e401.
- Khan, K.H.; Yap, T.A.; Yan, L.; Cunningham, D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin. J. Cancer 2013, 32, 253–265.
- Larsen, L.J.; Møller, L.B. Crosstalk of hedgehog and mTORC1 pathways. Cells 2020, 18, 2316.
- Jung, S.; Gámez-Díaz, L.; Proietti, M.; Grimbacher, B. Immune TOR-opathies," a novel disease entity in clinical immunology. Front. Immunol. 2018, 9, 966.
- Zhao, Y.; Sun, Y. Targeting the mTOR-DEPTOR pathway by CRL E3 ubiquitin ligases: Therapeutic application. Neoplasia 2012, 14, 360–367.
- Ebrahimi, S.; Hosseini, M.; Shahidsales, S.; Maftouh, M.; Ferns, G.A.; Ghayour-Mobarhan, M.; Hassanian, S.M.; Avan, A. Targeting the Akt/PI3K signaling pathway as a potential therapeutic strategy for the treatment of pancreatic Cancer. Curr. Med. Chem. 2017, 24, 1321–1331.
- Chen, S.; Chen, C.; Hu, Y.; Song, G.; Shen, X. The diverse roles of circular RNAs in pancreatic cancer. Pharmacol. Ther. 2021, 226, 107869.
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell. Mol. Life. Sci. 2015, 72, 2337–2347.
- Polivka, J., Jr.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175.
- Margaria, J.P.; Campa, C.C.; De Santis, M.C.; Hirsch, E.; Franco, I. The PI3K/Akt/mTOR pathway in polycystic kidney disease: A complex interaction with polycystins and primary cilium. Cell Signal 2020, 66, 09468.
- Mazzoletti, M.; Broggini, M. PI3K/AKT/mTOR inhibitors in ovarian cancer. Curr. Med. Chem. 2010, 17, 4433–4447.
- Duzgun, Z.; Eroglu, Z.; Biray Avci, C. Role of mTOR in glioblastoma. Gene 2016, 10, 187–190.
- Nowak, J.A. HER2 in colorectal carcinoma: Are we there yet? Surg. Pathol. Clin. 2020, 13, 485–502.
- Du, L.; Li, X.; Zhen, L.; Chen, W.; Mu, L.; Zhang, Y.; Song, A. Everolimus inhibits breast cancer cell growth through PI3K/AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 17, 7163–7169.
- Degan, S.E.; Gelman, I.H. Emerging roles for AKT isoform preference in cancer progression pathways. Mol. Cancer. Res. 2021, 19, 1251–1257.
- Baer, R.; Cintas, C.; Therville, N.; Guillermet-Guibert, J. Implication of PI3K/Akt pathway in pancreatic cancer: When PI3K isoforms matter? Adv. Biol. Regul. 2015, 59, 19–35.
- Wadhwa, B.; Makhdoomi, U.; Vishwakarma, R.; Malik, F. Protein kinase B: Emerging mechanisms of isoform-specific regulation of cellular signaling in cancer. Anticancer Drugs 2017, 28, 569–580.
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160.
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132.
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 15, 314–341.
- Selvarajoo, N.; Stanslas, J.; Islam, M.K.; Sagineedu, S.R.; Ho, K.L.; Lim, J.C.W. Pharmacological modulation of apoptosis and autophagy in the treatment of pancreatic cancer. Mini Rev. Med. Chem. 2022.
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J. Immunol. 2017, 1, 1006–1014.
- Briest, F.; Grabowski, P. PI3K-AKT-mTOR-signaling and beyond: The complex network in gastroenteropancreatic neuroendocrine neoplasms. Theranostics 2014, 29, 336–365.
- Tan, B.; Huang, Y.; Zhang, B.; Lin, N. The effect of ibrutinib on radiosensitivity in pancreatic cancer cells by targeting EGFR/AKT/mTOR signaling pathway. Biomed. Pharmacother. 2020, 128, 110133.
- Beyens, M.; Vandamme, T.; Peeters, M.; Van Camp, G.; Op de Beeck, K. Resistance to targeted treatment of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2019, 1, R109–R130.
- Hubbard, P.A.; Moody, C.L.; Murali, R. Allosteric modulation of Ras and the PI3K/AKT/mTOR pathway: Emerging therapeutic opportunities. Front. Physiol. 2014, 16, 478.
- Chen, H.; Zhou, L.; Wu, X.; Li, R.; Wen, J.; Sha, J.; Wen, X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front. Biosci. 2016, 1, 1084–1091.
- Wang, J.; Nishiyama, A.; Matsuyama, M.; Wang, Z.; Yuan, Y. The (pro)renin receptor: A novel biomarker and potential therapeutic target for various cancers. Cell Commun. Signal 2020, 6, 39.
- Nie, S.; Qian, X.; Shi, M.; Li, H.; Peng, C.; Ding, X.; Zhang, S.; Zhang, B.; Xu, G.; Lv, Y.; et al. ALDH1A3 accelerates pancreatic cancer metastasis by promoting glucose metabolism. Front. Oncol. 2020, 16, 10–915.
- Bao, B.; Wang, Z.; Li, Y.; Kong, D.; Ali, S.; Banerjee, S.; Ahmad, A.; Sarkar, F.H. The complexities of obesity and diabetes with the development and progression of pancreatic cancer. Biochim. Biophys. Acta. 2011, 1815, 135–146.
- Yan, X.; Hui, Y.; Hua, Y.; Huang, L.; Wang, L.; Peng, F.; Tang, C.; Liu, D.; Song, J.; Wang, F. EG-VEGF silencing inhibits cell proliferation and promotes cell apoptosis in pancreatic carcinoma via PI3K/AKT/mTOR signaling pathway. Biomed. Pharmacother. 2019, 109, 762–769.
- Su, C.C. Tanshinone IIA can inhibit MiaPaCa2 human pancreatic cancer cells by dual blockade of the Ras/Raf/MEK/ERK and PI3K/AKT/mTOR pathways. Oncol. Rep. 2018, 40, 3102–3111.
- Lin, S.; Pan, Y.; Xu, C. Effects of aspirin on pancreatic cancer cells PANC-1 and its potential molecular mechanism. J. Buon. 2020, 25, 2449–2455.
- Song, J.; Xu, J.; Guo, J.; Shang, Y.; Wang, J.; Wang, T. The enhancement of tetrandrine to gemcitabine-resistant PANC-1 cytochemical sensitivity involves the promotion of PI3K/Akt/mTOR-mediated apoptosis and AMPK-regulated autophagy. Acta. Histochem. 2021, 123, 151769.
- Singh, B.N.; Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem. Pharmacol. 2012, 1, 1154–1163.
- Liu, X.; Tan, X.; Liu, P.; Wu, Y.; Qian, S.; Zhang, X. Phosphoglycerate mutase 1 (PGAM1) promotes pancreatic ductal adenocarcinoma (PDAC) metastasis by acting as a novel downstream target of the PI3K/Akt/mTOR pathway. Oncol. Res. 2018, 23, 1123–1131.
- Zhou, B.; Xiang, J.; Zhan, C.; Liu, J.; Yan, S. STK33 promotes the growth and progression of human pancreatic neuroendocrine tumour via activation of the PI3K/AKT/mTOR pathway. Neuroendocrinology 2020, 110, 307–320.
This entry is offline, you can click here to edit this entry!