Aging is a progressive decline of an organism over time. In contrast, senescence occurs throughout an organism’s lifespan. It is a cell-cycle arrest preventing the proliferation of damaged cells. Cellular and molecular senescence timing is crucial for the pace of aging and disease development and progression. The accumulation of senescent cells during a lifespan leads to organismal senescence. Senescent multinucleated giant cells are present in many age-related diseases and cancer. Although senescence was assumed to be irreversible, studies now show that senescent multinucleated giant cells overcome senescence in various cancers, becoming the source of highly aggressive mononucleated stem-like cells, which divide and initiate tumor development and progression.
- aging
- senescence
- senescence-associated secretory phenotype
- SASP
- cell fusion
- multinucleation
- giant cells
- macrophages
- arteries
1. Introduction
Aging Versus Senescence
2. Giant Multinucleated Cell Formation
3. Giant Cells in Aging Arteries
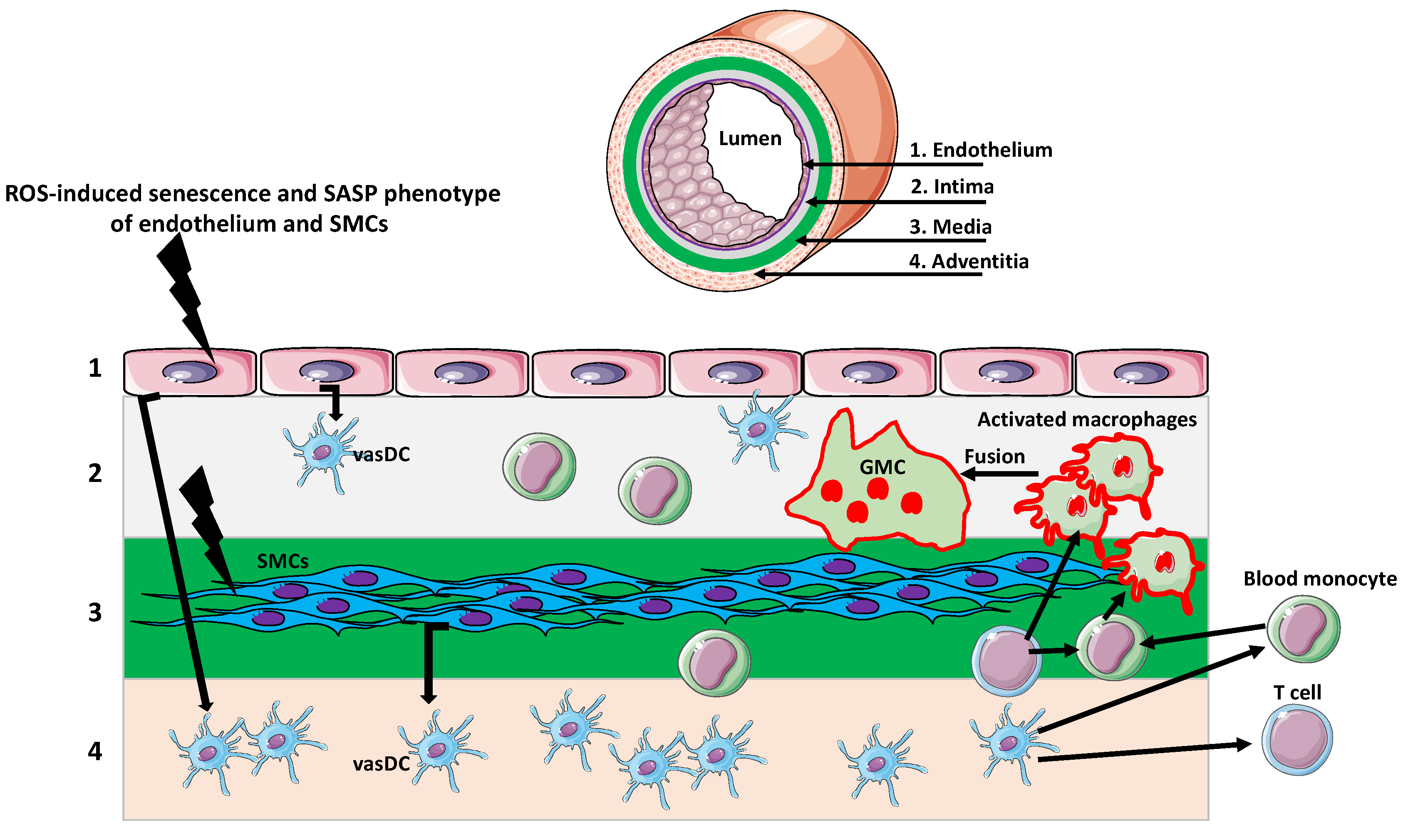
4. Giant Multinucleated Cells in Aging Gonads
This entry is adapted from the peer-reviewed paper 10.3390/biology11081121
References
- Terzi, M.Y.; Izmirli, M.; Gogebakan, B. The cell fate: Senescence or quiescence. Mol. Biol. Rep. 2016, 43, 1213–1220.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- Allsopp, R.C. Models of initiation of replicative senescence by loss of telomeric DNA. Exp. Gerontol. 1996, 31, 235–243.
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479.
- Zhang, R.; Chen, W.; Adams, P.D. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell Biol. 2007, 27, 2343–2358.
- Mooi, W.J.; Peeper, D.S. Oncogene-induced cell senescence-halting on the road to cancer. N. Engl. J. Med. 2006, 355, 1037–1046.
- Kohli, J.; Veenstra, I.; Demaria, M. The struggle of a good friend getting old: Cellular senescence in viral responses and therapy. EMBO Rep. 2021, 22, e52243.
- Wei, W.; Ji, S. Cellular senescence: Molecular mechanisms and pathogenicity. J. Cell Physiol. 2018, 233, 9121–9135.
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118.
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130.
- Yang, Y.; Shi, L.; Fu, X.; Ma, G.; Yang, Z.; Li, Y.; Zhou, Y.; Yuan, L.; Xia, Y.; Zhong, X.; et al. Metabolic and epigenetic dysfunctions underlie the arrest of in vitro fertilized human embryos in a senescent-like state. PLoS Biol. 2022, 20, e3001682.
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078.
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is Senescence Reversible? Curr. Drug Targets 2016, 17, 460–466.
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222.
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev. Biol. 2021, 9, 645593.
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77.
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 156–1576.
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485.
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22.
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118.
- Neurohr, G.E.; Terry, R.L.; Lengefeld, J.; Bonney, M.; Brittingham, G.P.; Moretto, F.; Miettinen, T.P.; Vaites, L.P.; Soares, L.M.; Paulo, J.A.; et al. Excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell 2019, 176, 1083–1097.e18.
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80.
- Dikovskaya, D.; Cole, J.J.; Mason, S.M.; Nixon, C.; Karim, S.A.; McGarry, L.; Clark, W.; Hewitt, R.N.; Sammons, M.A.; Zhu, J.; et al. Mitotic stress is an integral part of the oncogene-induced senescence program that promotes multinucleation and cell cycle arrest. Cell Rep. 2015, 12, 1483–1496.
- Radspieler, M.M.; Schindeldecker, M.; Stenzel, P.; Försch, S.; Tagscherer, K.E.; Herpel, E.; Hohenfellner, M.; Hatiboglu, G.; Roth, W.; Macher-Goeppinger, S. Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol. Lett. 2019, 18, 2654–2660.
- Vergnes, L.; Péterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin B1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433.
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716.
- De Oliveira Mann, C.C.; Kranzusch, P.J. cGAS conducts micronuclei DNA surveillance. Trends Cell Biol. 2017, 27, 697–698.
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674.
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686.
- Victorelli, S.; Passos, J.F. Reactive Oxygen Species Detection in Senescent Cells. Methods Mol. Biol. 2019, 1896, 21–29.
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347.
- Cory, S.; Huang, D.C.S.; Adams, J.M. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 8590–8607.
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190.
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522.
- Kloc, M.; Subuddhi, A.; Uosef, A.; Kubiak, J.Z.; Ghobrial, R.M. Monocyte-Macrophage Lineage Cell Fusion. Int. J. Mol. Sci. 2022, 23, 6553.
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590.
- Watanabe, R.; Hashimoto, M. Aging-Related Vascular Inflammation: Giant Cell Arteritis and Neurological Disorders. Front Aging Neurosci. 2022, 14, 843305.
- Mohan, S.V.; Liao, Y.J.; Kim, J.W.; Goronzy, J.J.; Weyand, C.M. Giant cell arteritis: Immune and vascular aging as disease risk factors. Arthritis Res. Ther. 2011, 13, 231.
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867.
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314.
- Kloc, M.; Ghobrial, R.M. Chronic allograft rejection: A significant hurdle to transplant success. Burns Trauma. 2014, 2, 3–10.
- Weyand, C.M.; Liao, Y.J.; Goronzy, J.J. The immunopathology of giant cell arteritis: Diagnostic and therapeutic implications. J. Neuroophthalmol. 2012, 32, 259–265.
- Jiemy, W.F.; van Sleen, Y.; van der Geest, K.S.; Ten Berge, H.A.; Abdulahad, W.H.; Sandovici, M.; Boots, A.M.; Heeringa, P.; Brouwer, E. Distinct macrophage phenotypes skewed by local granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) are associated with tissue destruction and intimal hyperplasia in giant cell arteritis. Clin. Transl. Immunol. 2020, 9, e1164.
- Briley, S.M.; Jasti, S.; McCracken, J.M.; Hornick, J.E.; Fegley, B.; Pritchard, M.T.; Duncan, F.E. Reproductive age-associated fibrosis in the stroma of the mammalian ovary. Reproduction 2016, 152, 245–260.
- Foley, K.G.; Pritchard, M.T.; Duncan, F.E. Macrophage-derived multinucleated giant cells: Hallmarks of the aging ovary. Reproduction 2021, 161, V5–V9.