Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The low water solubility of pharmacoactive molecules limits their pharmacological potential, but the solubility parameter cannot compromise, and so different approaches are employed to enhance their bioavailability. Pharmaceutically active molecules with low solubility convey a higher risk of failure for drug innovation and development. Pharmacokinetics, pharmacodynamics, and several other parameters, such as drug distribution, protein binding and absorption, are majorly affected by their solubility.
- solubility
- bioavailability
- dissolution
- nanoparticles
1. Introduction
The solubility of the drug, the solution, and its gastrointestinal permeability are essential factors that control the amount of absorption and absorption speed, along with the bioavailability of the drug [1]. A necessary factor involved in absorption after a drug’s oral administration is the aqueous solubility of therapeutics. The drug solubility is the dissolution rate at which the drug molecule or the dosage form allows for entering the solution, and it is essential when the time of dissolution is restricted [2]. However, the drug’ bioavailability depends on water solubility, dissolution rate, drug permeability, susceptibility to efflux mechanisms, and first-pass metabolism [3]. “Solubility” has been well-defined as the quantity of solute, which dissolves in a quantity of solvent. Quantity refers as the concentration of the solute in a saturated solution at a definite temperature. The solubility has been represented through multiple concentration expressions—for example, parts, percentage, molality, molarity, volume fraction, and mole fraction. Qualitatively, solubility can be termed as a spontaneous collaboration between two substances to create a homogenous dispersion at the molecular level. The solute can be referred to as at equilibrium with the solvent in a saturated solution [4][5][6].
In recent years, according to drug detection, the number of poorly soluble drugs has increased, with 70% of novel medications presenting low aqueous solubility [7]. The controlling factors for the in vivo bioavailability of oral formulations of these drugs are the low solubility and low dissolution rate in gastrointestinal solutions. Thus, an essential issue relating to drug development has been recognized as being in vitro dissolution and increasing the speed of dissolving low solvable drugs in addition to improving their bioavailability, representing a major task for pharmaceutical experts. [8][9].
For the medicinal product to be immersed, it must exist in a water-soluble state at the place of absorption [10][11][12]. The solubility and permeability represent promising factors for in vivo absorption. They can be improved through solubility enhancement techniques [13].
Rebamipide belongs to BCS class IV drugs. It exhibits poor bioavailability and has difficulties in formulation preparation for oral administration. Due to its limitations, it is impossible to formulate a self-nano emulsifying drug delivery system (SNEDDS) formulation using this substance. To enhance the solubility of this drug, a SNEDDS formulation was prepared by complexing rebamipide with its counter ion. Tetra-butyl phosphonium hydroxide (TBPOH) and NaOH were used as counter ions to prepare a complex. Okawa et al. reported that the complexes prepared with rebamipide, Reb–TBPOH complex and Reb–NaOH complex, showed enhanced solubility and absorption in in vitro as well as in in vivo studies. Asper recent scenario of pharmaceutical developing market Figure 1 demonstrates the development of different formulations and their ratio as per BCS classification.
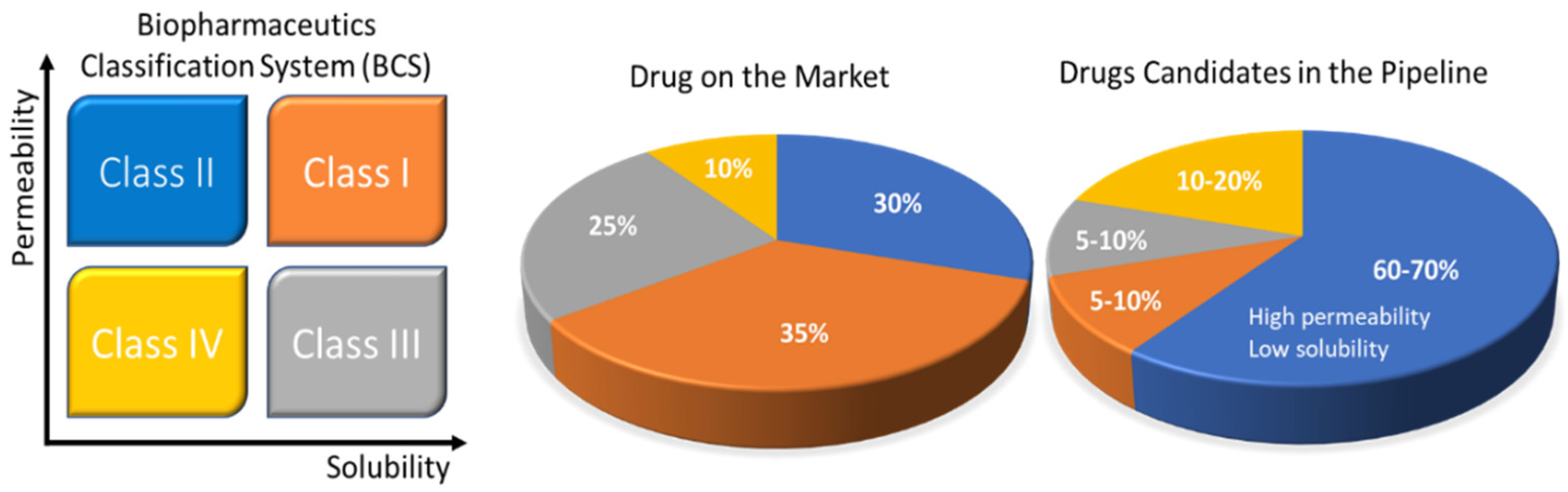
Figure 1. Solubility challenges that plague the oral drug-delivery frontier. The Biopharmaceutics Classification System (BCS) uses drug permeability and solubility as metrics for oral absorption. The four categories include BCS Class I (orange: high solubility, high permeability), Class II (blue: low solubility, high permeability), Class III (black: high solubility, low permeability), and Class IV (yellow: low solubility, low permeability). The pie charts to the right show the estimated distribution of marketed and pipeline drugs by BCS classes. The pharmaceutical company data are also presented in the pie charts. Reprinted and adapted with permission from ref. [14]. Copyright 2018 Bioconjugate chemistry.
Quercetin is a highly hydrophobic drug, which is a polyphenolic flavonoid. It finds application as an antioxidant. It exhibits anti-proliferative and chemopreventive characteristics. It was found to have potential against colon, lung, ovarian and breast cancer but, due to its poor solubility and low bioavailability, its pharmacological effects are limited. Kakran et al. used bottom-up and top-down approaches as side reduction techniques to prepare nanoparticles [15]. High-pressure homogenization and bead milling approaches were applied top-down and EPN-evaporative precipitation of nanosuspension was applied as a bottom-up technique to prepare quercetin nanoparticles with enhanced solubility and bioavailability.
Johnson et al. developed seventeen types of poly(N-isopropyl acrylamide) (PNIPAM)-based excipients differing in molar mass and with a variety of end groups. These polymers were studied for their capability to enhance the water solubility of phenytoin. To improve the solubility of phenytoin, Johnson et al. synthesized three polymeric excipients: PNIPAM, poly(N,N-dimethyl acrylamide) (PDMAm) and PHEAmpoly(N-hydroxyethyl acrylamide) (PHEAm). Synthesized polymers were investigated for their solubility enhancement effect on phenytoin. Johnson et al. described a detailed understanding of the critical importance of PNIPAM for the solubility enhancement of phenytoin [16][17].
Chen et al. worked on the drug solubility enhancement of docetaxel (DTX). Docetaxel finds application as a chemotherapeutic agent for cancer treatment. Chen prepared three types of inclusion complex among docetaxel and H1-3 (ethylenediamine modified beta cyclodextrins) with ethylene, propylene and butylene parts. Chen proved that the complexation of DTX with H1-3 is an effective tactic to enhance the solubility and prepare a less toxic and highly active DTX formulation. This approach can maximize its clinical applicability in cancer treatment [18].
In recent decades, solid dispersion technology was extensively studied to develop an amorphous carrier to increase the bioavailability, solubility and dissolution rate of drugs with poor water solubility. The solid dispersion preparation methodology and selection of appropriate carriers will play a critical role in its biological behavior [19]. Here are some techniques to prepare amorphous solid dispersions to enhance the bioavailability, solubility and therapeutic efficacy of drugs: (a) cryogenic processing techniques, (b) freeze drying, (c) fluid-bed coating, (d) spray drying, (e) microwave irradiation, (f) co-precipitation method, (g) electrostatic spinning, (h) supercritical anti-solvent (SAS), (i) HME technique, (j) MeltrexTM, (k) melt agglomeration, and the (l) KinetiSolVR dispersing (KSD) technique.
The drug solubility is mainly influenced by its chemical arrangement and the conditions of its solution. The molecular assembly defines its molecular volume, crystal energy, hydrogen bonding, ionizability, and lipophilicity, which determines the drug solubility. pH, additives, time, temperature, cosolvents, and ionic strength will affect the solution conditions. Pharmaceutical compounds with poor water solubility can intensely decrease output in drug discovery and development.
A “good compound” must reach the target site’s inactive focuses. Solubility of compound affects their absorption, permeability, and potency. Highly potent and permeable compounds are suitable for low aqueous solubility.
2. Biopharmaceutical Classification System
BCS is one of the most applied scientific classification systems of drug substances based on their permeability and solubility as described in Figure 2. There are two significant factors which regulate the speed and scope of oral drug absorption that is aqueous solubility and intestinal permeability [20].

Figure 2. Biopharmaceutical classification.
The compounds of drugs can be categorized into four classes, according to BCS.
When the maximum potency of the drug is soluble in ≤250 mL of the aqueous standard, it means drugs are highly soluble, and their range of pH is 1.0 to 7.5; otherwise, the drug elements are measured as not very soluble. Biopharmaceutical researchers are continuously trying to make a biologically mimicking system that can match conditions such as gut pH, food content, and peristalsis to predict in vivo performance precisely. Several biopharmaceutical research developments were made between 1960 and 1970; numerous studies were carried out and established the relationship between the effect of dissolution and formulation parameters on drug bioavailability. For the proper evaluation of any formulation’s dissolution rate, the first dissolution test apparatus was introduced in 1970, USP apparatus I (basket type), and afterwards, another USP Apparatus II (paddle type) was introduced [21]. Using this apparatus, it has been possible to predict the in vivo performance of the formulation from the in vitro tests. However, because the in vivo performance of every formulation depends on several variables, improvements have been made to the in vivo performance of dosage forms. Figure 3 provides a chronological list of some of the significant studies conducted in this area.
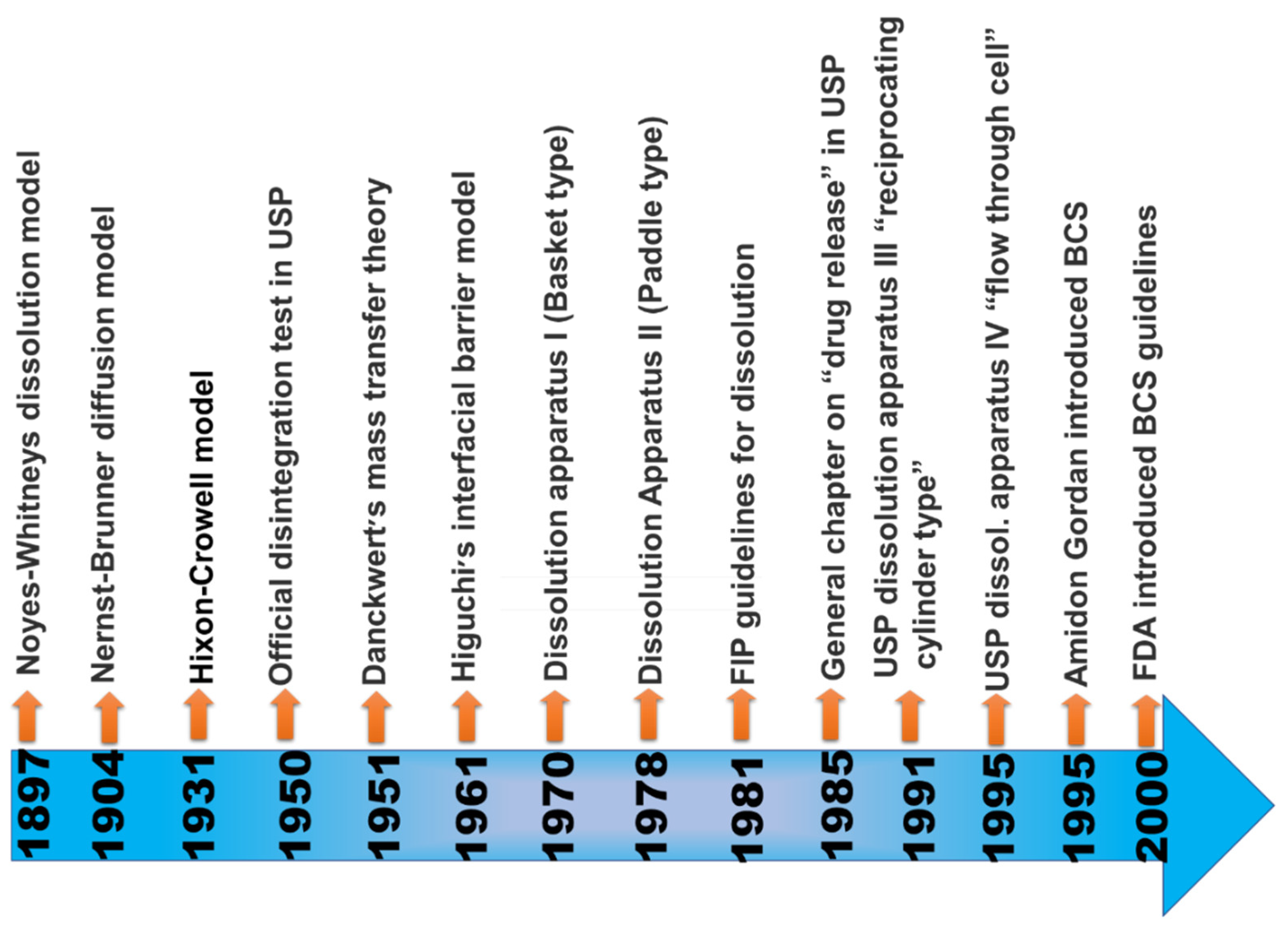
Figure 3. Evolutionary history of biopharmaceutical classification system [22].
3. Importance of Solubility Enhancement
The main difficulty in the advancement of a new chemical entity is low aqueous solubility. For oral drugs, the most rate-limiting factor is solubility and reaching its concentration aspirated in full circulation for the pharmacological response. To obtain an approved concentration providing the necessary pharmacological reaction, solubility is one of the essential parameters [23]. Hydrophobic drugs usually require high doses and need high dosage regimens to influence therapeutic plasma concentrations after administration [24].
The oral administration of drugs is the most accessible and generally applied way of delivering drugs thanks to flexible administration, profitability, high compliance by the patient, flexibility in the dosage design, and fewer sterility restrictions. For this reason, most generics companies prefer to yield bioequivalent oral pharmaceuticals [3]. However, the main problem with oral administration is related to its dosage form design and its low bioavailability.
The factors affecting oral bioavailability include drug permeability, first-pass metabolism, dissolution rate, aqueous solubility and susceptibility to efflux mechanisms. The most common reasons for low oral bioavailability are associated with low permeability and poor solubility.
In the case of other dosage forms such as parenteral formulations, solubility plays a critical role [25]. Drugs with poor water solubility frequently require a high dosage of the drug to extend therapeutic plasma concentrations following oral administration. The essential criteria include being in an aqueous medium at the absorption site for the absorption of any pharmacological compound. H2O is the preferred solvent for preparing liquefied pharmaceutical preparations. A drug’s slow solubility in aqueous media is the foremost difficulty for formulation scientists [26]. Most drugs are weakly basic or weakly acidic, having less water solubility. Almost 40% of NCEs (new chemical entities) are insoluble in water, as discovered in the pharmaceutical sector.
Drug solubility enhancement is the most challenging factor in the field of drug discovery. In the literature, several strategies exist and have been described for the solubility improvement of poorly water-soluble drugs. These techniques are preferred based on specific features such as the characteristics of the drug under consideration, intended dosage form types, and chosen excipient characteristics.
The low dissolution rate and low solubility in the aqueous gastrointestinal fluids lead to inadequate bioavailability. Mainly aimed at group II compounds conferring to the BCS, bioavailability can be improved through enhancing the dissolution rate and solubility of the drug in gastrointestinal fluids. The ratio restrictive factor for the class II BCS is the release of the drug from the dosage form and gastric fluid solubility and not the absorption, and thus enhancing the solubility will improve the bioavailability of BCS class II drug molecules [3][26][27].
The negative impact of drugs having poor solubility involves less bioavailability and poor absorption for IV dosing, due to that high cost (frequent high-dose administration) are challenges [25].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10092055
References
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res 1995, 12, 413–420.
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499.
- Krishnaiah, Y.S. Pharmaceutical technologies for enhancing oral bioavailability of poorly soluble drugs. J. Bioequiv. Availab. 2010, 2, 28–36.
- Nidhi, K.; Indrajeet, S.; Khushboo, M.; Gauri, K.; Sen, D.D.J. Hydrotropy: A promising tool for solubility enhancement: A review. Int. J. Drug Dev. Res. 2011, 3, 26–33.
- Martin, A.; Bustamanate, P.; Chun, A.H.C. Physical Pharmacy; BI Wavely Pvt. Ltd.: New Delhi, India, 1994; Volume 4, p. 223.
- Gennaro, A.R. Remington’s Pharmaceutical Sciences, 17th ed.; Mack Publishing Co.: Easton, PA, USA, 1985.
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10.
- Hu, J.; Johnston, K.P.; Williams, R.O., 3rd. Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Dev. Ind. Pharm. 2004, 30, 233–245.
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133.
- Curry, S.H. Applied biopharmaceutics and pharmacokinetics, Leon Shargel and Andrew BC Yu, Appleton-Century-Crofts, New York, 1980. Biopharm. Drug Dispos. 1982, 3, 287–290.
- Sinko, P.J. Martin’s Physical Pharmacy and Pharmaceutical Sciences; Lippincott Williams & Wilkins: Philadelphia, PA, USA; London, UK, 2005.
- Carstensen, J.T. Pharmaceutical Preformulation; Technomic Publishing Company: Lancaster, PA, USA, 1998; Volume 14, p. 47.
- Brahmankar, D.M.; Jaiswal, S.B. Biopharmaceutics and Pharmacokinetics: A Treatise; Vallabh Prakashan: Delhi, India, 2005.
- Ting, J.M.; Porter III, W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in polymer design for enhancing oral drug solubility and delivery. Bioconjug. Chem. 2018, 29, 939–952.
- Kakran, M.; Li, L.; Müller, R.H. Overcoming the challenge of poor drug solubility. Pharm. Eng. 2012, 32, 82–89.
- Johnson, L.M.; Li, Z.; LaBelle, A.J.; Bates, F.S.; Lodge, T.P.; Hillmyer, M.A. Impact of Polymer Excipient Molar Mass and End Groups on Hydrophobic Drug Solubility Enhancement. Macromolecules 2017, 50, 1102–1112.
- Johnson, L.M.; Hillmyer, M.A. Critical Excipient Properties for the Dissolution Enhancement of Phenytoin. ACS Omega 2019, 4, 19116–19127.
- Chen, X.-Y.; Yang, H.-W.; Chi, S.-M.; Yue, L.-L.; Ruan, Q.; Lei, Z.; Zhu, H.-Y.; Zhao, Y. Solubility and biological activity enhancement of docetaxel via formation of inclusion complexes with three alkylenediamine-modified β-cyclodextrins. RSC Adv. 2021, 11, 6292–6303.
- Alshehri, S.; Imam, S.S.; Hussain, A.; Altamimi, M.A.; Alruwaili, N.K.; Alotaibi, F.; Alanazi, A.; Shakeel, F. Potential of solid dispersions to enhance solubility, bioavailability, and therapeutic efficacy of poorly water-soluble drugs: Newer formulation techniques, current marketed scenario and patents. Drug Deliv. 2020, 27, 1625–1643.
- Papich, M.G.; Martinez, M.N. Applying Biopharmaceutical Classification System (BCS) Criteria to Predict Oral Absorption of Drugs in Dogs: Challenges and Pitfalls. AAPS J. 2015, 17, 948–964.
- Bai, G.; Armenante, P.M.; Plank, R.V.; Gentzler, M.; Ford, K.; Harmon, P. Hydrodynamic Investigation of USP Dissolution Test Apparatus II. J. Pharm. Sci. 2007, 96, 2327–2349.
- Shekhawat, P.B.; Pokharkar, V.B. Understanding peroral absorption: Regulatory aspects and contemporary approaches to tackling solubility and permeability hurdles. Acta Pharm. Sin. B 2017, 7, 260–280.
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. Int. Sch. Res. Not. ChemInform 2012, 42, 195727.
- Varandal, A.B.; Magar, D.; Saudagar, R. Different approaches toward the enhancement of drug solubility: A review. J. Adv. Pharm. Educ. Res. 2013, 3, 415–426.
- Di, L.; Kerns, E. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015.
- Sharma, D.; Soni, M.; Kumar, S.; Gupta, G. Solubility enhancement—Eminent role in poorly soluble drugs. Res. J. Pharm. Technol. 2009, 2, 220–224.
- Kumar, A.; Sahoo, S.K.; Padhee, K.; Kochar, P.S.; Sathapathy, A.; Pathak, N. Review on solubility enhancement techniques for hydrophobic drugs. Pharm. Glob. 2011, 3, 1–7.
This entry is offline, you can click here to edit this entry!