Mitochondria is an organelle with a double membrane and it is very widely distributed, presenting in almost all cells of the body [
1]. Regarding the origin of mitochondria, the endosymbiotic theory is widely accepted by researchers. In this theory mitochondria are said to be derived from the merger of bacteria with proto-eukaryotic cells [
2]. Mitochondria are critical energy-supplying organelles whose primary function is to produce adenosine triphosphate (ATP). They provide energy to the cell through the tricarboxylic acid cycle (TCA cycle) and oxidative phosphorylation. However, there is no doubt that mitochondria have several other roles in the cell, such as regulation of calcium homeostasis, production of reactive oxygen species, cell proliferation and metabolism. All these functions are important for the normal life activities of cells [
3]. Meanwhile, mitochondria are also important for immune cells, including T cells, macrophages, and neutrophils [
4]. According to recent studies, mitochondrial damage may be related to the pathogenesis of rheumatoid arthritis [
1].
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease that mainly affects synovial joints and causes a series of severe inflammatory reactions [
5]. The etiology of RA is complex and still being explored. Although recent therapeutic breakthroughs have been made, there has been no cure for the disease until now. Co-epitopes of human leukocyte antigen DR (HLA-DR) alleles in major histocompatibility complex II (MHC-II) are important genetic factors in RA [
6]. It has also been hypothesized that the onset of RA is associated with the dysregulation of immune signaling pathways. The patient’s autoantibodies incorrectly recognize and attack self-antigens, leading to a series of inflammatory responses in the joints [
7]. Autoantibodies such as rheumatoid factor, anti-citrullinated protein antibodies (ACPA) and anti-carbamylated protein antibodies are present in high levels in the plasma of RA patients [
8]. Moreover, RA affects approximately 0.1–2.0% of the global population, with a higher incidence in females than in males [
9]. The main symptoms of RA are swelling and pain in the joints and, in severe cases, deformity of the joints. This can be accompanied by complications such as anemia, osteoporosis, cardiovascular disease, and lung disorders [
10].
Mitochondrial dysfunction is a critical factor in developing autoimmune diseases, including RA. Mitochondria are involved in disease pathogenesis by acting on different signaling pathways through direct and indirect effects.
2. Three Ways That Mitochondrial Dysfunction Leads to RA
In RA, the most severely damaged cells are chondrocytes and synovial cells. If the mitochondrial function in these two types of cells is disrupted, it will lead to the dysfunction of cell physiology, and then trigger RA. Chondrocytes are a mature cell type in cartilage. They have a significant role in the production and maintenance of the cartilage matrix, which is essential for the health of the human joint. At the same time, mitochondria are important in maintaining the normal function of chondrocytes and the stability of the vivo environment [
3].The synovial membrane is found in the inner lining of the synovium and is composed of fibroblast-like synovial cells (FLS) and macrophage-like synovial cells (MLS) [
11]. RA is generally accompanied by the development of synovitis. In RA, synovial cells are activated due to stimulation and FLS secretes cytokines (e.g., Interleukin-6) to initiate inflammation at the joint. Activated FLS are hyperproliferative and resistant to apoptosis, leading to an increase in their number and exacerbating the symptoms of arthritis [
12]. Many studies have shown that mitochondrial dysfunction is associated with abnormalities in synovial cell function, including promotion of synovial cell inflammation, inhibition of synovial cell apoptosis, promotion of synovial cell invasiveness, and promotion of synovial cell proliferation [
11].
2.1. Abnormal Energy Metabolism
Mitochondria are indispensable for the energy supply of chondrocytes due to their essential physiological function of producing ATP from glucose. Glucose is converted to pyruvate in the cytoplasm through glycolysis. Then, pyruvate enters the mitochondrial matrix and is converted to acetyl coenzyme A which undergoes Krebs cycle and provides NADH for the electron transport chain [
13]. The electron transport chain consists mainly of four enzyme complexes, coenzyme Q and cytochrome C, which transfer electrons and pump out hydrogen ions. The potential energy provided by the outflow of hydrogen ions powers the synthesis of ATP by ATPase. Because of the action of ATPase, one molecule of glucose can eventually produce 32 ATP [
14]. Oxidative phosphorylation is of significant importance for cellular energy supply and is a highly efficient way of ATP synthesis (
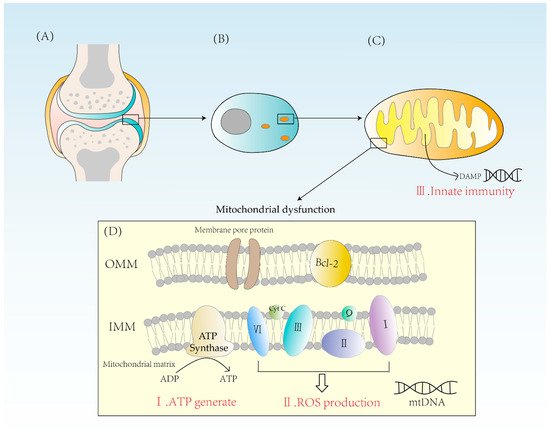
Figure 1).
Figure 1. (A) A diagram of the articular cartilage structure. Cartilage is mainly composed of chondrocytes. (B) The distribution of mitochondria in chondrocytes is related to the energy requirement. (C) Mitochondrion is a double-membrane organelle. Mitochondria have two membranes, an outer membrane and an inner membrane. Mitochondrial inner membrane folds to form cristae. The mitochondrial matrix contains mtDNA. DAMPs activate innate immunity, causing inflammatory reactions. (D) Mitochondria synthesize ATP through ATPase and form ROS through complexes I, II, III, and IV. When mitochondria are dysfunctional, both excessive ROS and energy imbalance will contribute to the occurrence of RA. OMM = outer mitochondrial membrane, IMM = inner mitochondrial membrane, Bcl-2 = B-cell leukemia/lymphoma-2, DAMPs = damage-associated molecular patterns.
If mitochondrial dysfunction occurs, the balance between glycolysis and oxidative phosphorylation is disrupted, affecting the synthesis of ATP, which in turn has irreversible effects on the production and metabolism of various substances. This is especially so for chondrocytes because, as the only mature cell in cartilage, if their ATP synthesis is reduced it will not only negatively affect the function of the chondrocyte itself but also have some impact on the extracellular protein synthesis and stability of the matrix. Meanwhile, ATP deficiency due to mitochondrial dysfunction may disrupt the repair of cartilage degradation [
3].
2.2. Excess ROS Production
One of the reactive oxygen species (ROS) sources in chondrocytes is the mitochondria. The reactive oxygen species synthesized in mitochondria, also known as mtROS, is mainly generated due to proton leakage in the electron transport chain. In normal conditions, the production of one oxygen molecule requires the transfer of four electrons by the ETC after complex IV and four hydrogen ions are combined to form two H2O molecules (
Figure 1) [
15].
Though the electrons in the ETC leak during transfer, they will stay in unstable positions and are susceptible to oxidation by adjacent oxygen, producing ROS and hydroxyl (OH-) [
16]. Under normal circumstances, about 2% of oxygen is used to produce reactive oxygen species (ROS), which has important physiological implications in the cell. A sufficient amount of ROS is required for chondrocyte repair and apoptosis, cytokine production, and extracellular matrix synthesis [
17,
18]. A rise in ROS levels leads to severe damage to cellular structures, including DNA, lipids, and proteins. Generally speaking, the mitochondrial antioxidant defense system can scavenge excess O2- and hydrogen peroxide to keep ROS levels low. At the same time, mitochondria can scavenge ROS produced by other cellular sources (e.g., macrophages) in order to protect cells from oxidative damage [
3].
2.3. Activation of Innate Immunity
Mitochondria are essential organelles in the synthesis of ATP and control of programmed cell death in synovial cells, creating ROS [
11]. They play an important role in energy supply, cell cycle control, and cell metabolism. Further, mitochondria also have a solid immunological function. Mitochondria are formed from internalized bacteria, according to the endosymbiotic theory. When mitochondria are damaged, damage-related molecular patterns (DAMPs) are released into the cytoplasm. These endogenous molecules can bind to corresponding receptors to elicit an immune response (
Figure 1). For example, mtDNA released from mitochondria during mitochondrial damage is known as DAMP, which triggers immune responses and causes inflammation. Therefore, factors leading to mtDNA leakage may cause mitochondrial disorder, and eventually lead to the occurrence of inflammatory reactions, which may ultimately lead to RA [
19]. DAMPs can activate innate immune cells and cause innate immune response to a certain extent. At present, a variety of DAMPs have been found, mainly including high-mobility histone B1, IL-1α, heat shock protein, etc. The process of inducing immune responses includes NF-κB signaling and induces the production of pro-inflammatory cytokines such as tumor necrosis factor-β (TNF-β) [
20]. In this way, a strong innate immunity is activated, leading to the development of RA.
3. Effects of Mitochondrial Dysfunction on Immune Cells in RA
3.1. T Cell
Under normal physiological conditions, primary T cells transform, after recognizing antigens on antigen-presenting cells (APCs), into effector T cells and begin to proliferate. Effector T cells have a mitochondrial oxidative metabolism dependency [
1]. Glycolysis provides energy for the proliferation of effector T cells, as well as substrates for DNA and protein synthesis. Therefore, mitochondria play an important role in T cell proliferation and function [
21].
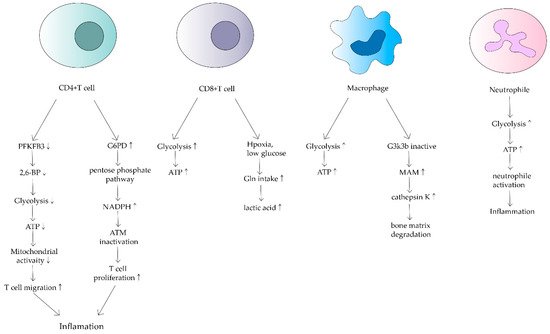
CD4+ T cells are the main cellular component of synovitis in RA and a key driver of pathogenic immunity. Yang Z et al. found that ATP levels, oxygen consumption and lactate production were reduced in CD4+ T cells of RA patients [
22]. They suggested that in RA patients, 6-phosphofuran-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), an essential rate-limiting enzyme of the glycolytic pathway is inhibited. Under normal physiological conditions, PFKFB3 produces fructose 2,6-bisphosphate (a metabotropic activator of the glycolytic enzyme PFK-1), which contributes to glycolysis [
23]. However, in CD4+ T cells in RA, induction of PFKFB3 is inhibited, resulting in a decreased glycolysis rate and a reduction in ATP synthesis [
24]. Meanwhile, glucose-6-phosphate dehydrogenase (G6PD) expression is upregulated in CD4+ T cells. G6PD can catalyze the pentose phosphate pathway (PPP), causing NADPH and glutathione concentrations to increase (
Figure 2). Besides, glutathione has an antioxidant effect. The enhanced antioxidant effect of excess glutathione prevents the activation of cell cycle checkpoint kinase ataxia telangiectasia-mutated (ATM), providing the possibility of T cell over proliferation. Meanwhile, overproliferating T cells are biased towards differentiation into inflammatory Th1 and Th17, exacerbating the response [
25].
Figure 2. Effects of mitochondrial dysfunction on different immune cells in RA. This shows that ATP synthesis increases in CD4+ T cells, macrophages and neutrophils. In contrast, it decreases in CD8+ T cells. Different cells have different mechanisms that lead to inflammation and bone matrix degradation, causing the development of RA.
In addition to the adverse effects of reduced ATP production by oxidative phosphorylation in mitochondria on T cells, the TCA cycle of CD4+ T cells in RA can be disrupted by disruptions that increase levels of upstream metabolites such as α-ketoglutarate, citric acid and acetyl coenzyme A, leading to loss of granulomatous activity inline T cells [
26]. This metabolic disruption enhances T-cell motility and migration, leading to greater inflammation and susceptibility to inflammation [
27].
CD8+ T cells in RA synovium are also affected. However, in contrast to CD4+ T cells, CD8+ T cells in RA have been shown to increase the uptake of glutamine in hypoxic and low glucose conditions, resulting in lactate production increases(
Figure 2) [
28]. Alterations in both metabolic pathways of CD4+ T cells and CD8+ T cells can increase inflammatory mediators and are associated with RA development.
3.2. Macrophage
RA also alters the metabolic phenotype of macrophages, which is mainly related to the modulation of the normal activity of mitochondria. Glycolysis and oxidative phosphorylation are upregulated in macrophages due to pro-inflammatory requirements(
Figure 2) [
29]. In macrophages in RA patients, because of the upregulation of glycolysis and oxidative phosphorylation, more oxygen is consumed and more ATP is produced. Tight junctions with the endoplasmic reticulum are established to form mitochondria-associated membranes (MAMs) [
30]. MAMs promote calcium transfer to maintain mitochondrial activity. In turn, the increased MAM association is dependent on the inactivation of glycogen synthase kinase 3b (GSK3b), which acts as a metabolic switch to regulate cellular respiration. In RA, most macrophages can inactivate GSK3b, allowing for increased MAM association, facilitating calcium transfer and increased ATP synthesis in mitochondria. In contrast, the formation of MAM and the persistence of GSK3b inactivation produces the collagenase cathepsin K, which makes it an important component of bone resorption and metabolism [
31]. Cathepsin K destroys bone joints through the enhancement of bone resorption, something which is closely associated with the development of RA [
32,
33].
3.3. Neutrophil
Neutrophils are the source of stimulation and damage to extracellular mitochondrial DNA in autoimmune diseases. Mitochondrial dysregulation in neutrophils has an important impact on the course of RA [
34]. Neutrophils are the origin of autoantigens which can promote inflammation and lead to tissue damage. The degree of neutrophil activity is heavily dependent on how much energy is produced by glycolysis. Researchers have found that neutrophils from RA synovial fluid exhibit enhanced glycolytic gene expression compared to peripheral blood neutrophils from other parts of the body (
Figure 2). Mitochondria produce large amounts of ATP when they are dysfunctional, thus neutrophil activity is enhanced, leading to a series of inflammatory responses. Although a relevant role in RA has not been demonstrated, mitochondrial dysfunction has been shown to affect neutrophils, such as chemotaxis [
34]. Because of the relatively few relevant studies, mitochondrial metabolism of neutrophils in RA deserves further investigation.