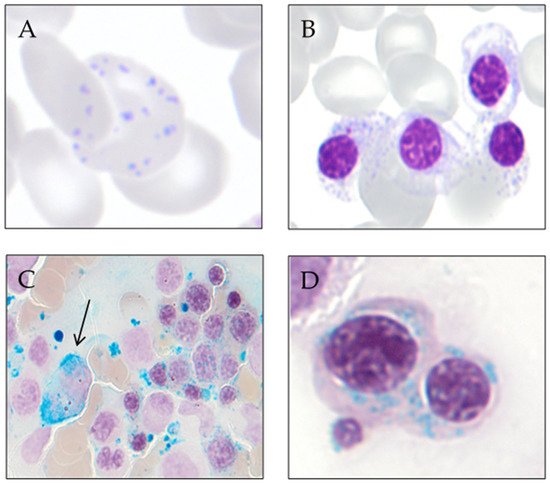
Ring sideroblasts are erythroblasts with an abnormal accumulation of iron in their perinuclear mitochondria, and their presence defines sideroblastic anemias. To reveal them, it is necessary to apply Prussian blue stain (Perls’ reaction) to bone marrow aspirate smears. Ring sideroblasts are found in a variety of pathological conditions, both congenital and acquired. Among the acquired causes of sideroblastic anemias, we can find clonal and non-clonal disorders.
2. Historical Context
In 1942, Hans Grüneberg demonstrated, using the Prussian blue staining, the presence of free iron in the cytoplasm of some erythroblasts (sideroblasts) and in some mature erythrocytes (siderocytes) [
2]. In 1945, Cooley [
3] described a patient with sex-linked anemia, which probably corresponded to a case of the nonsyndromic form of X-linked SA (XLSA), since later Cotter et al. identified mutations in
ALAS2 and ringed sideroblasts in the same family [
4]. In 1956, Björkman described a series of four patients with chronic refractory anemia with numerous abnormal bone marrow sideroblasts, one of which developed leukemia. This is probably the first description of myelodysplastic syndromes with ring sideroblasts [
5]. Sideroblastic anemias were recognized as a specific subtype of anemia in the 1960s [
6]. Within the last 30 years, with the important advances in molecular biology, the genetic origin of more than two-thirds of congenital sideroblastic anemias cases, and a great proportion of cases of acquired clonal disease have been clarified.
3. Ring Sideroblast Definition
Prussian blue staining (Perls’ reaction) is an essential technique in the study of patients with anemia. Its application in bone marrow aspirate smears permits the analysis of macrophage iron storage and the assessment of the number and characteristics of sideroblasts. This stain reveals ferritin granules within the erythroblasts and hemosiderin in bone marrow macrophages [
7].
A proportion of normal erythroblasts exhibit few (1–5) iron-containing granules randomly distributed around the cytoplasm. Such erythroblasts are designated as sideroblasts and stand for 20–50% of the erythroblasts in a normal bone marrow [
8]. Sideroblasts are visible in bone marrow aspirate smears but not in bone marrow biopsy sections since erythroblastic iron is lost in bone marrow biopsy processing [
7]. Ring sideroblasts are aberrant sideroblasts where iron-loaded mitochondria are visualized by Prussian blue staining as a perinuclear ring of green-blue granules. There have been various definitions of a ring sideroblast causing confusion and controversy among clinicians [
9]. The International Working Group on Morphology of Myelodysplastic Syndrome (IWGM-MDS) defined three types of sideroblasts [
10]:
- -
-
Type 1 sideroblasts: < 5 siderotic granules in the cytoplasm.
- -
-
Type 2 sideroblasts: ≥ 5 siderotic granules, but no perinuclear distribution.
- -
-
Type 3 or ring sideroblasts: ≥ 5 siderotic granules in a perinuclear position, covering at least one-third of the nuclear circumference.
To establish the percentage of sideroblasts in a bone marrow, at least 100 erythroid precursors of all maturation stages should be counted. This definition of ring sideroblast proposed by the IWG-MDS was incorporated into the 2008 and updated in the 2017 edition of the WHO classifications of Tumours of Haematopoietic and Lymphoid Tissues [
7,
11].
4. Ring Sideroblast Formation
The discovery of genetic variations underlying ring sideroblast has led to a better understanding of the pathophysiology of the sideroblastic anemias. Nevertheless, our understanding of how ring sideroblasts arise is limited. There are many open questions in this regard: Are they detrimental to the erythroblast? Are they a cause or a consequence?
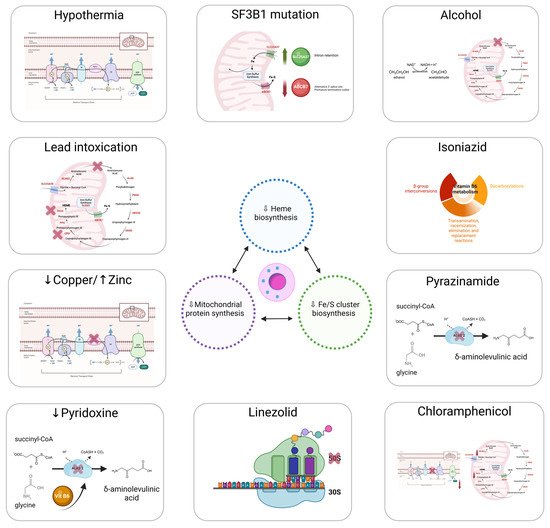
Mitochondrion is the epicenter of sideroblastic anemia. Disrupted mitochondrial metabolism is present among all sideroblastic anemias for which an etiology has been defined. The mitochondrial functions affected in sideroblastic anemias are Heme biosynthesis; iron–sulfur cluster (ISC) biogenesis; and synthesis of mitochondrial proteins, general proteins, or proteins dedicated to oxidative metabolism. All of these defects lead to an aberrant accumulation of iron in the mitochondria of erythroblasts [
1,
12].
Figure 1 represents the main causes of acquired sideroblastic anemia.
Figure 1. Main causes of acquired sideroblastic anemia.
Mitochondria provide the majority of the ATP needed by eukaryotic cells through oxidative phosphorylation [
13]. The adult erythrocyte is the only mammalian cell that does not have mitochondria, relying exclusively on anaerobic glycolysis for ATP production [
14]. Mitochondria are semi-autonomous organelles that most likely evolved from free-floating prokaryotes that infiltrated eukaryotic cells over a billion years ago [
15].
The mitochondria genome is small, around 16 kb, and replicates autonomously conserving vestiges of their prior self-sufficiency [
16,
17]. Mitochondrial DNA, along with several bacterial genomes, displays an intron-free circular structure [
18]. Chromatin absence and a limited DNA-repair capacity enable mutations in the mitochondrial DNA to develop sideroblastic anemia [
19].
Replication within mitochondria occurs independently of the nuclear genome [
20]. Mitochondria are stochastically distributed to progeny after cells undergo mitosis. As a result, acquired mitochondrial abnormalities are passed on in an unequal manner to the daughter cells. This feature is important to some of the hereditary mitochondrial disorders that produce sideroblastic anemia. This characteristic also presents a conundrum regarding acquired sideroblastic anemias. Some cases of sideroblastic anemia linked with myelodysplasia include mutations that prevent some cytochromes from working properly [
21,
22]. It remains uncertain how mitochondria with deteriorated enzymatic performance become so prevalent in cells. Reasonably, impairment of the mitochondrion should not confer a survival advantage.
4.1. Heme Synthesis
Most of congenital sideroblastic anemias are due to Heme deficiency. Heme is a critical component of several mitochondrial enzymes (cytochromes b, c1, c, a, and a3), as well as cytosolic enzymes such as catalase [
23]. Heme plays structural and functional roles as an essential member in the hemoglobin structure. Particularly, Heme regulates the translation of globin mRNA, mediates reversible oxygen binding [
24], and stabilizes the globin protein chains.
Heme biosynthesis initiates with the condensation of glycine and succinyl-CoA to generate 5′-aminolevulinic acid (ALA) [
25], consuming pyridoxal phosphate (active form of vitamin B6) as a cofactor in the reaction [
26]. ALA then is transported to cytoplasm, where, after numerous enzymatic reactions, it is converted to coproporphyrinogen III [
27]. This molecule again reaches the mitochondrion, where it undergoes further modifications and has iron inserted into the protoporphyrin IX ring by ferrochelatase (FECH), eventually generating Heme [
28]. Porphyria is caused by defects in the cytoplasmic phases of Heme production. For instance, functional anomalies of the enzyme porphobilinogen deaminase produce acute intermittent porphyria [
29]. Only 10 patients with erythropoietic protoporphyria (EPP) [
30], a disorder characterized by pronounced deficiency of FECH, have been reported to present ring sideroblasts [
31].
Aminolevulinic acid synthase (ALAS) is both the first and the rate-limiting enzyme in Heme biosynthesis [
25]. Heme regulates its activity by inhibiting feedback. The two ALAS genes have been cloned and allocated to specific chromosomal regions. The
ALAS-1 (also known as
ALAS-n) gene has been localized to chromosome 3 (3p21) [
32], being highly expressed in the liver. ALAS-1 maintains steady levels, providing basal Heme production required by all cells. ALAS-1 is a key member in the Heme biosynthetic process in mammalian cells, with the exception of erythroid cells, where erythroid-specific 5-aminolevulinate synthase (ALAS-2 or ALAS-E) governs the initial stage of Heme biosynthesis [
33]. This enzyme is encoded by a gene on the X chromosome (Xp11.21), and its expression is restricted to the erythroid lineage [
34]. Expression and activity of ALAS-2 is regulated by iron levels, as well as Heme-mediated feedback regulation [
35]. Importantly, deficiency of ALAS-2 accounts for around 40% of all congenital sideroblastic anemia cases [
36].
4.2. ISC Biogenesis
Iron–sulfur clusters (ISCs) are core components of many mitochondrial and extramitochondrial proteins, showing catalytic activity [
37]. ISC plays a fundamental role in cellular iron uptake regulation, iron storage, Heme synthesis, and interaction with iron regulatory protein 1 (IRP1) and FECH [
38]. Congenital sideroblastic anemia, accompanied by defects in the transfer stage of ISC biogenesis, has been reported.
4.3. Mitochondrial Respiratory Complex Proteins and Mitochondrial Protein Synthesis
A broad defect in mitochondrial protein synthesis has been described to lead to congenital sideroblastic anemias associated with neuromuscular disease and lactic acidosis consequent to impaired mitochondrial energy metabolism.
The homeostasis of iron is vital to human health, and iron dyshomeostasis can lead to various disorders since excess iron can promote the generation of deleterious reactive oxygen species (ROS).
Iron homeostasis is maintained by iron regulatory proteins (IRP1 and IRP2) and the iron-responsive element (IRE) signaling pathway [
39].
Intracellular iron is used for multiple functions; if not utilized, it is stored in ferritin or exported by ferroportin in order to maintain the labile iron pool within narrow limits to avoid toxicity. Erythroblasts are cells specialized in iron uptake, and more than 80% of this iron is directed to mitochondria [
40,
41].
While normal erythroblasts store their iron in cytosolic ferritin, which is encoded by the
FTH1 and
FTL genes, ring sideroblasts store their iron in mitochondrial ferritin (FtMt), which is encoded by the FTMT gene, an intronless gene located on chromosome 5q23.1 [
42]. FtMt contains ferroxidase activity; thus, it is likely to sequester potentially damaging free iron [
43]. Ultimately, overexpression of FtMt results in mitochondrial iron loading and cytosolic iron deficiency [
44].
The nature of this iron and the fact that these cells survive this massive overload has long been a conundrum. Bessis and Breton-Gorius found by electron microscopy that this electron dense iron gave images similar to those of the iron cores of ferritin and proposed that it was ferritin [
45]. At that time, the structural complexity of ferritin was not known, and there was then no molecular basis for mitochondrial targeting. Different studies showed that there is little, if any, FtMt in normal erythroblasts but very high levels in the iron-loaded mitochondria in ringed sideroblasts [
46,
47].
Through Fenton chemistry (Equation (1)), iron catalyzes the creation of reactive oxygen species [
48]. Molecules such as the hydroxyl radical (−OH) form in environments where oxidation processes take place around iron [
49]. The mitochondrion’s oxidative metabolic machinery facilitates a suitable setting to produce reactive oxygen species. In sideroblastic anemia, the main damage that results in iron-laden mitochondria might trigger a feedback cycle of aggravating mitochondrial impairment [
50]. For instance, (−OH) stimulates the peroxidation of lipids and proteins, as well as the formation of cross-links in DNA strands. Given the previously suggested lack of DNA-repair enzymes in mitochondria, the latter event might be extremely harmful.
Equation (1) is the Fenton reaction. The Fenton reaction involves iron II (Fe
2+) reacting with H
2O
2 to yield a hydroxy radical (OH) and a hydroxide ion (OH
−):