2. Viral Ιnfections as Τriggers for Parkinsonism and PD Development
Several studies have demonstrated that viruses may contribute to the etiology of PD and parkinsonism, despite the fact that the underlying molecular and cellular mechanisms remain obscure. The first recorded association between viral infections and parkinsonism was observed during the Spanish flu and the appearance of encephalitis lethargica, an unknown disease with parkinsonian phenotype in survivors [
41]. Major human viruses, such as hepatitis C virus (HCV) [
42], herpes simplex virus-1 (HSV-1) [
43], human immunodeficiency virus (HIV) [
44], varicella-zoster virus (VZV) [
45], West Nile virus (WNV) [
46], Japanese encephalitis virus (JEV) [
47,
48], and Epstein–Barr virus (EBV) [
49], have all been cited as risk factors for PD development or parkinsonism [
3]. Notably, the role of influenza A virus (IAV) in the etiology of the transient parkinsonian phenotype [
50] and in PD development [
3] has been documented in several in vivo and especially in vitro studies. A case-control study found that an influenza diagnosis was linked to PD development 10 years following infection onset [
51], while IAV was found postmortem in the substantia nigra of PD patients [
52]. Furthermore, H5N1 infection in a mouse model resulted in Parkinson’s phenomenology, sustained microglial activation, and α-syn aggregation, leading to dopaminergic neuron loss in SNpc [
53]. Similarly, H1N1 infection in mice resulted in persistent microglial activation as a sign of chronic virus-induced neuroinflammation that could potentially lead to neurodegeneration [
54]. More recently, another in vitro study has demonstrated that H1N1 replication can directly disrupt protein homeostasis, inducing α-syn aggregates in Lund human mesencephalic dopaminergic cells, but failing to regulate TAR DNA-binding protein 43 (TDP-43) or tau protein. Those results clearly hint at a selective effect of H1N1 virus on α-syn misfolding [
55].
The key pathophysiological processes by which viruses contribute to parkinsonism development remain unclear; however, direct neuronal damage, sustained neuroinflammation, cerebral edema due to virus-mediated damage of brain endothelium, and induction of α-syn aggregation have all been proposed as crucial neurobiological pathways of dopaminergic neuron loss and α-syn pathology [
3]. Notably, due to its tendency to entrap viral particles and reduce viral replication, α-syn has been postulated to be a natural antiviral defense mechanism for neurons [
56]. This notion was supported by in vivo experiments, where WNV-infected α-syn-knockout mice showed decreased survival compared to the control group [
57]. Additionally, it has been suggested that viruses can cause α-syn aggregation and oligomerization through molecular mimicry mechanisms [
58,
59]. Taken together, these observations strongly support the notion that virus-mediated neuronal deposition of pathological α-syn may induce neurotoxicity and PD pathology.
The relationship between other members of the human
Coronaviridae family, such as OC43 and 229E, and PD has been previously described, since antibodies against these coronaviruses were found in the cerebrospinal fluid (CSF) of PD patients [
60]. The novel coronavirus SARS-CoV-2 emerged in China at the end of 2019 and triggered an outbreak of atypical viral pneumonia [
61]. Due to its enhanced transmissibility, this unusual coronavirus disease, also known as coronavirus disease 2019 (COVID-19), marched fast over the world, constituting a huge public health burden [
62,
63]. SARS-CoV-2 spreads via infected secretions, such as saliva and respiratory droplets, through direct, indirect, or close contact with infected patients, even if COVID-19 symptomatology is absent [
64,
65]. While symptoms of COVID-19 are primarily systemic or respiratory, several studies demonstrate the presence of a broad spectrum of neuropsychiatric consequences including anosmia, ageusia, altered consciousness, headache, seizures, and paresthesias [
66,
67,
68]. Several studies have shown that COVID-19-related neurological sequelae might persist long after the acute phase of infection [
69]. The term “long” or “post”-COVID-19 syndrome refers to a syndrome observed after the acute infection period and it is characterized by the presence of a combination of COVID-19-related symptoms lasting for more than 12 weeks [
70]. These symptoms cannot be explained by an alternative diagnosis and are considered a disability under the Americans with Disabilities Act (ADA) [
71]. The post-COVID-19 syndrome includes a plethora of neurological manifestations such as fatigue, brain fog, cognitive impairment, and olfactory dysfunctions [
72,
73,
74], many of which are also present in PD [
2]. Thus, since SARS-CoV-2 shares immunopathological similarities with other viruses linked to parkinsonism, such as influenza [
75], and because of COVID-19-related neurological consequences, it is reasonable to suspect that these persistent symptoms might be a prologue to a post-COVID-19 new-onset neurological disease.
3. SARS-CoV-2 Infection and PD Overlaps
3.1. Clinical Co-Manifestations
To date, only few cases of parkinsonism have been reported in literature following COVID-19 infection [
19,
20,
76,
77,
78]. In these studies, the authors speculate a possible causative link between COVID-19 infection and a post-COVID new-onset parkinsonian phenotype, but they do not address the possibility of prodromal, pre-symptomatic PD, which became symptomatic as a result of biological or psychological stress processes associated with COVID-19. In the latter case, SARS-CoV-2 infection could act as a trigger that unmasks an underlying PD phenotype, possibly by stimulating neuroinflammatory and neurodegenerative cascades. In addition, SARS-CoV-2 infection has been demonstrated to significantly worsen motor and non-motor symptoms in people with pre-existing PD [
79,
80]. Considering the prevalence of post-COVID-19 syndrome [
81,
82], a multicenter study found that 23 out of 27 PD patients developed post-COVID-19 symptoms, with the most common long term effects of COVID-19 being the deterioration of motor function and the requirement for increased levodopa daily dose, followed by fatigue, cognitive disturbances including brain fog, and sleep disorders [
83].
Probably the clinical symptoms most commonly shared between PD and COVID-19 are gustatory and especially olfactory dysfunctions. Indeed, both olfactory and gustatory impairments are among the earliest non-motor PD features [
84,
85]. Surprisingly, these are common early onset symptoms of COVID-19 and it has been observed that hyposmia–anosmia and dysgeusia could persist long after viral load decline, constituting a key clinical manifestation of the long COVID-19 syndrome [
86,
87]. Due to lack of evidence regarding the definite CNS infiltration, the olfactory route is discussed as a way for SARS-CoV-2 to gain access to the CNS. Indeed, a postmortem study demonstrated that the highest levels of SARS-CoV-2 RNA and spike protein (S protein) among various brain areas were found in the olfactory mucosal–nervous milieu, as well as in neuroanatomical areas related to the olfactory tract. In this regard, the olfactory mucosa could serve as an “anatomical bridge” for SARS-CoV-2 CNS invasion through axonal transport [
88]. Furthermore, angiotensin-converting enzyme 2 (ACE2), an essential cell surface receptor responsible for S protein-mediated entry of SARS-CoV-2, was found to be expressed by epithelial cells of the human olfactory mucosa [
89]. The extent of α-syn pathology in other brain regions has been substantially linked with the pathological burden in the olfactory bulb, suggesting that PD pathology extends along olfactory pathways [
90]. The Braak hypothesis proposes that LB are initially found in olfactory structures, such as the olfactory bulb, and then they gradually spread towards the brain stem and ultimately to the cerebral cortex, strengthening the scenario that the earliest lesions could develop at non nigral areas [
91,
92]. Accordingly, Beach and colleagues have demonstrated that the olfactory bulb constitutes a primary affected area in α-synucleinopathies, including PD. In fact, it was suggested that the extent of α-synucleinopathy in the olfactory bulb strongly predicts the neuropathological confirmation of PD and reflects the severity of α-synucleinopathy in other brain regions [
93]. Based on these studies, one could hypothesize that the olfactory route might pose a way for SARS-CoV-2 to gain access to the CNS, where it can modify neuropathological pathways pertinent to PD development.
Another common pathology shared between PD and COVID-19 is the deregulation and dysfunction of the gastrointestinal (GI) tract. GI symptoms and intestinal inflammation may emerge years before clinical indications of PD become apparent [
94,
95]. Specifically, gastrointestinal dysbiosis has been proposed to be involved in PD pathogenesis [
96] and the enteric nervous system has been previously identified as a primary region for abnormal α-syn aggregation, which may then spread from the periphery to the CNS [
97,
98,
99]. Specifically, the dorsal motor nucleus of the vagus nerve (DMV) receives signals from vagal parasympathetic neurons that project to the entire GI system. The DMV is involved in the PD–neuroanatomical pathway, since a monosynaptic nigro–vagal pathway that connects the SNpc to the DMV has been identified in the rat [
100]. In postmortem PD studies, the DMV and the vagus nerve itself are among the most frequently afflicted structures [
101,
102] and they constitute principal areas of LB accumulation, even at the earliest stages of disease development [
91]. In vitro research has shown that pathological α-syn may spread from the gut to the brain through the vagus nerve, with DMV being the first area of the brain to be impacted. From there, α-syn can spread to other PD brain regions including the SNpc, resulting in dopaminergic neuron loss and the appearance of the parkinsonian phenotype [
103]. Interestingly, the vagus nerve has been proposed as a pathway through which SARS-CoV-2 can retrogradely invade the CNS, thus enhancing its neuroinvasiveness [
104,
105].
Importantly, other GI manifestations, such as diarrhea, emerged as common clinical symptoms of COVID-19, while SARS-CoV-2 RNA detection in fecal samples may persist post-infection [
106]. On top of that, gut microbiota imbalance due to extrapulmonary SARS-CoV-2 infection has also been observed in COVID-19 [
107,
108]. This warrants further investigation because GI microbiota equilibrium plays an important role in several physiological processes ensuring brain integrity and neurogenesis [
109,
110]. Taken together, the above observations suggest that SARS-CoV-2 infection could promote PD development and progression through a virus-exerted dysfunction of the GI system.
3.2. Inflammatory and Molecular Overlapping Pathways
Common inflammatory events unraveling during PD development and observed in the acute phase of SARS-CoV-2 infection, as well as after COVID-19 remission, may indicate a link between these two disorders. Virus-mediated sustained or aberrant neuroinflammation could be a decisive pathobiological process for the initiation of a neurodegenerative disease, such as PD, long after recovery from the viral infection [
111,
112,
113]. Indeed, growing evidence indicates that SARS-CoV-2 induces neuroinflammation [
114] through its neurotropic, neuroinvasive, and neurovirulence effects [
115,
116] or even via immune-mediated pathways [
117]. SARS-CoV-2 infection also triggers systemic inflammatory responses and induces cytokine release [
118]. Severe COVID-19 is characterized by a cytokine storm syndrome, which is a major cause of mortality [
118,
119]. Several studies have demonstrated the presence of inflammatory mediators, such as increased levels of pro- and anti-inflammatory interleukins (IL-1, IL-2, IL-6, IL-10) and tumor necrosis factor-alpha (TNF-α) in the serum of COVID-19 patients [
120,
121,
122,
123]. Interestingly, a small prospective observational study had previously found that high levels of IL-6 were linked to a higher chance of developing PD [
124]. Evidently, an exacerbated systemic infection that causes a huge release of inflammatory mediators, including cytokines, chemokines, and antibodies, could lead to increased blood–brain barrier (BBB) permeability [
125]. Functional and structural integrity of the BBB is pivotal in maintaining brain homeostasis [
126]. A neurovascular unit (NVU) consists of multiple cell types, including brain microvascular endothelial cells (BMVECs), astrocytes, pericytes, microglia, and neurons, connected together with extracellular matrix components, and is a rigorous regulator of BBB permeability [
127]. NVU disruption has been previously associated with neurodegenerative diseases [
128]. In particular, BMVECs constitute an important component of NVU and are intricately interconnected through tight junction (TJ) proteins. However, inflammation affects BBB integrity and stability mainly through cytokine-induced degradation of TJ proteins [
129]. SARS-CoV-2-mediated brain endothelial inflammation, upregulation of inflammatory mediators, and most significantly, disruption of BBB stability, have also been observed in human BMVECs [
130]. According to in vitro studies, SARS-CoV-2 was shown to infect human BMVECs and cause a decrease in TJ protein expression [
130,
131]. Furthermore, incubation of human BMVECs with S protein resulted in enhanced ACE2 expression, thereby facilitating viral entry and inducing neuroinflammation [
132].
When BBB becomes impaired, pro-inflammatory cytokines and factors, innate immune cells from the periphery, and SARS-CoV-2 could possibly pass through and infiltrate the CNS. In that case, the CNS professional immune cells, microglia and astrocytes, may also become activated [
133,
134]. Neuroinflammation is then likely to set in fast, leading to elevated production of cytokines, chemokines, reactive oxygen species (ROS), and secondary messengers [
135]. Microglia, which are highly susceptible to pro-inflammatory stimuli, are concentrated in areas harboring dopaminergic neurons, making them particularly vulnerable to inflammatory mediators [
136,
137]. Interestingly, the S1 subunit of S protein was found to efficiently trigger neuroinflammation, including microglia activation, release of multiple pro-inflammatory cytokines, and cause behavioral deficits in rats [
138]. Consequently, these neuroinflammatory cascades lead to enhanced apoptotic activity, increased ROS levels, mitochondrial dysfunction, and eventually neurodegeneration [
139,
140].
Finally, cellular senescence is a core homeostatic event that provides yet another, age- and state-dependent substrate for neurodegeneration and the development of diseases like AD and PD [
141,
142]. Cellular senescence in the aging brain affects both neuronal and non-neuronal cells, and it is characterized by a broad array of interconnected disruptions, such as disruptions in autophagy, bioenergetics, and mitochondrial dynamics, as well as the onset of low-grade inflammation [
142]. This cumulative array of dysfunction culminates in the accumulation of proteopathic seeds, including tau, amyloids, and α-syn, and tissue-wide remodeling [
141]. It has been shown that SARS-CoV-2 infection induces “immunosenescence” and enhances the senescence-associated secretory phenotype (SASP) in infected tissues, via disruption of host antiviral mechanisms, such as interferon signaling pathways [
143,
144,
145]. Taken together, all the aforementioned studies strongly indicate that the COVID-19 cytokine storm and innate immunity dysregulation may cause neuroinflammation and, in consequence, neurodegeneration.
Neuropathological findings in postmortem brain tissues from COVID-19 patients further support the involvement of COVID-19-related neuroinflammatory processes in PD development. A postmortem brain study of 43 COVID-19 patients has shown activation of microglia and CNS infiltration by cytotoxic T-lymphocytes, more apparent in the brainstem [
146]. Regardless of COVID-19 disease severity, significant inflammatory responses such as astrogliosis, microglia activation, and perivascular T-lymphocyte infiltration were observed postmortem in both white and gray matter of patient brains [
147]. Performing single-nucleus RNA sequencing and immunohistochemistry on tissue from a group of individuals who died with COVID-19 and a group of individuals who died from other causes, Yang and colleagues revealed glia transcriptomic changes that indicated a COVID-19-associated activation of inflammatory pathways. The ensuing dysregulation of homeostatic pathways could potentially lead to neurodegeneration [
148]. Specifically, microglia and astrocytic subpopulations were enriched by inflammatory genes and deregulated neuroprotective ones that had been previously linked to PD and other human neurodegenerative diseases, such as the glial fibrillary acidic protein (GFAP), the interferon-induced transmembrane protein-3 (IFITM3), and others [
149,
150].
Another mechanism that may contribute to PD pathogenesis involves the renin–angiotensin system and ACE2, which are implicated in the pathophysiology of COVID-19 and may play a role in neuroinflammation-mediated neurodegeneration in PD [
151,
152]. ACE2 is highly expressed in several brain areas [
153], including striatum [
154], the substantia nigra, the olfactory bulb [
155], and the brain endothelium [
130,
156,
157]. Induced pluripotent stem cells (IPCS) derived from midbrain dopaminergic neurons were shown to be vulnerable to SARS-CoV-2 infection in vitro [
158], unravelling the potentially direct neurotrophic effect of SARS-CoV-2 in strategic PD areas. Furthermore, SARS-CoV-2-induced Toll-like receptor (TLR) overactivation led to ACE2 upregulation and promoted the neurotrophic and neuroinflammatory outcomes of SARS-CoV-2 infection [
159]. TLRs belong to the family of innate immune receptors and play an important role in the activation of innate immunity, including activation of glial cells. TLR-mediated stimulation of intracellular signaling pathways culminates in the release of proinflammatory mediators such as IL-6, IL-1, TNF-a, and nuclear factor-κB (NF-κB) [
160]. Protein-to-protein interaction between SARS-CoV-2 S protein and TLR-4 has been previously recorded [
161]. SARS-CoV-2-mediated overactivation of the TLRs may lead to hyperinflammation, ACE2 upregulation and microglia switching from the neuroprotective to the neurotoxic phenotype [
159,
162]. In sequel, sustained gliosis and prolonged neuroinflammation could lead to α-syn aggregation and finally loss of dopaminergic neurons in the SNpc [
112].
Aside from neuroinflammation, dysregulation of several homeostatic molecular pathways has been identified in PD onset and development. These alterations also occur during host–virus interactions as the virus attempts to direct critical cellular infrastructure towards completion of its own lifecycle. SARS-CoV-2 viral proteins were shown to post-translationally reconfigure the biological function of 24 host proteins expressed in lung. The latter act as perturbators and interact with 44 CNS proteins that are known to be implicated in PD pathogenesis [
163]. Specifically, SARS-CoV-2-mediated deregulation of Rab7a and nucleoporin-62 (NUP62) could be strongly involved in PD pathogenesis, because Rab7 lysosomal protein decreases α-syn aggregation and associated neurotoxicity [
164], while NUP62 is crucial for autophagosome development [
165]. Furthermore, SARS-CoV-2 proteins can interact and bind to a variety of human protein trafficking molecules. Protein trafficking, translation, transcription, and ubiquitination regulation are all coordinated by these biomolecules, leading to neuroprotection, protection of BBB integrity, and neurogenesis [
166]. A recent study demonstrated a direct interaction between SARS-CoV-2 nucleocapsid protein (N-protein) and α-syn, which led to the aggregation of the latter into amyloid fibrils, a highly pathogenic form of the protein, linked to PD. Co-administration of SARS-CoV-2 N protein and α-syn to a PD cell model resulted in twice the neuron loss due to neurotoxicity compared to control cells treated with α-syn alone [
167].
Other important cellular processes implicated in the loss of dopaminergic neurons in SNpc are thought to be oxidative stress and mitochondrial dysfunction, endoplasmic reticulum stress, and the impairment of protein degradation systems [
168,
169,
170].
A key molecular factor in PD development and progression is mitochondrial dysfunction and oxidative stress [
171,
172]. An imbalance between ROS generation and cellular antioxidant activity leads to oxidative stress and ROS can further affect mitochondria, attenuating adenosine triphosphate (ATP) production as well as causing damage to mitochondrial DNA [
173]. In addition to causing direct cellular damage, oxidative stress can speed up neuron degeneration by inducing inflammatory or apoptotic pathways, such as NF-κB or caspase activation [
174]. In PD studies, mitochondrial dysfunction may occur months before the onset of striatal dopaminergic neuron loss [
175] and PD patients have been well documented to possess reduced or deficient mitochondrial complex I activity in the SNpc [
176,
177]. In mice, accumulation of wild-type α-syn in dopaminergic neurons reduced mitochondrial complex I activity and elevated ROS production, leading to cell death [
178]. SARS-CoV-2 seems to interact with and manipulate mitochondria in order to hijack and evade mitochondria-mediated immune response for its own replication and survival [
179,
180]. In this effort, SARS-CoV-2 may induce mitochondrial impairment [
181,
182], mitochondria-mediated oxidative stress, and mitochondrial damage through mitochondrial membrane depolarization, mitochondrial permeability transition pore opening, and enhanced ROS release [
183,
184,
185]. Furthermore, the virus prevents mitophagy by blocking the binding of p62 and microtubule-associated protein 1A/1B-light chain 3 (LC3), thereby hindering viral RNA breakdown [
185].
Finally, mitochondria aid the antiviral immune response by allowing release of pro-inflammatory cytokines [
186]. ACE2 has been suggested to regulate mitochondrial function [
187]. Its expression is decreased when SARS-CoV-2 S protein binds to ACE2 on microglia cells, causing ATP reduction and activation of the ROS-generating enzyme NADPH oxidase [
188]. The ensuing increase in ROS production and oxygen consumption may lead to neuroinflammation and loss of neighbor dopaminergic neurons [
189].
Endoplasmic reticulum (ER) stress has been linked to neurodegenerative diseases, including PD [
190,
191]. ER homeostasis disruption and extended ER stress lead to misfolded protein accumulation and may stimulate particular proapoptotic pathways through the activation of the transcription factor C/EBP homologous protein (CHOP) and cysteine proteases caspase-4/12 [
192,
193]. Growing evidence suggests that SARS-CoV-2 proteins interact with the ER compartment and may induce ER stress [
194,
195]. SARS-CoV-2 open reading frame 8 (ORF8) is capable of inducing ER stress by triggering the activating transcription factor 6 (ATF6) and inositol-requiring enzymes 1 (IRE1) branches of the ER stress pathway [
196], potentially leading to α-syn accumulation [
197]. Aside from initiating apoptotic pathways, ER stress is a powerful stimulator of NF-κB activation and inflammatory gene transcription [
198,
199]. SARS-CoV-2 also appears to activate NF-κB, causing inflammation, possibly through ER stress or via interaction with the non-structural protein Nsp5 [
200]. Notably, NF-κB is a crucial transcription factor that regulates inflammation and dopaminergic neurons loss in PD patients [
201]. Hence, deregulation of this signaling pathway has been linked to PD onset and pathology [
202] by favoring α-syn accumulation, aggregation, and spreading, oxidative stress-induced neuron apoptosis, neuroinflammation, and dopaminergic neuron loss [
139,
203,
204].
When aggregation and deposition of misfolded α-syn elicit dopaminergic neuron loss, protein degradation systems come to the rescue. The ubiquitin–proteasome system (UPS) and the autophagy–lysosomal pathway (ALP) are important proteolytic systems in neurons and critical for refolding or elimination of misfolded proteins; therefore, they play a significant role in cellular homeostasis [
205]. Impairment or even failure of these systems may contribute to PD pathogenesis and progression [
21,
206]. SARS-CoV-2 virulent components, such as ORF proteins, seem to modify autophagy formation and function, leading to SARS-CoV-2-induced autophagy disruption and potentially neuron damage [
207,
208]. Specifically, ORF3a was shown to impede autophagosome–lysosome (A-L) fusion and ALP formation by interacting directly with the VPS39 subunit of the homotypic fusion and protein sorting (HOPS) complex. ORF3a further damages lysosomes and impairs their function. Remarkably, this feature of HOPS-VPS39-mediated A-L fusion inhibition appears to be unique to SARS-CoV-2, since the quite similar ORF3a of SARS-CoV was ineffective in inhibiting A-L fusion [
209]. Furthermore, another study found that although ORF7a protein stimulates autophagy, it also limits A-L fusion progression by downregulating the SNAP29 protein via caspase 3 (CASP3) activation, providing a mechanism through which SARS-CoV-2 uses the autophagic system to facilitate its own propagation [
210]. Interestingly, a SARS-CoV-2 papain-like protease has been identified to directly cleave serine/threonine unc-51-like kinase (ULK1) and prevent ULK1-ATG13 complex formation [
211]. ULK1 is an upstream autophagy orchestrator, which phosphorylates key regulatory proteins in autophagosome formation [
212]. In this regard, ULK1 cleavage is expected to completely inhibit the ALP function, due to lack of autophagosome formation. Evidently, autophagy is crucially involved in the regulation of the antiviral immune response. The striking correlation between SARS-CoV-2-induced aberrant inflammation and the observed autophagy defects [
213] suggests that the virus-induced cytokine storm could be mediated by the failure of autophagy mechanisms to maintain cellular homeostasis.
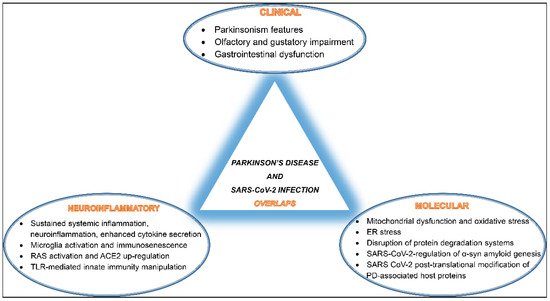
Overall, SARS-CoV-2 seems to interfere and disrupt several host cellular and molecular pathways involved in proper neuronal functions, potentially promoting PD pathogenesis. A summary of these overlaps is depicted in (Figure 1).
Figure 1. A schematic diagram of SARS-CoV-2 infection and Parkinson’s disease (PD) development overlaps listing shared clinical manifestations, common neuroinflammatory events, and mutually activated molecular pathways.