Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Cancer therapy-related cardiopulmonary dysfunction (CTRCPD) is one of the most undesirable side effects of cancer therapy and leads to limitations to cancer treatment. Chemoradiation therapy or immunotherapy promote acute and chronic cardiopulmonary damage by inducing reactive oxygen species, DNA damage, inflammation, fibrosis, deregulation of cellular immunity, cardiopulmonary failure, and non-malignant related deaths among cancer-free patients who received cancer therapy. The efficacy of the radiation depends on the degree of DNA damage, which results in cancer cell death.
- chemoradiation
- immunotherapy
- reactive oxygen species
- antioxidants
1. Fibroblast Activation and Cardiopulmonary Inflammation in Response to Cancer Therapy
Generally, fibroblast cells are undifferentiated, activated by a variety of signaling molecules such as platelet-derived growth factors (PDGF) and TGF- β, which are differentiated into myofibroblasts [1]. The differentiated myofibroblasts interact with cardiomyocytes and pulmonary tissues to promote the molecular pathogenesis of fibrosis and the production of the extracellular matrix (ECM). The conversion of fibroblasts to myofibroblasts enables the contraction of the ECM to cover the open wound [2]. Fibroblasts play a vital role in the inflammation and immune cell recruitment at injured sites. Additionally, fibroblasts secrete inflammation responsive cytokines and play a role in the inflammation and immune cell recruitment to wounded sites to clear dead cells and scars. Furthermore, fibroblasts produce inflammation responsive cytokines and chemokines, including TNF-α, TGF-β, IL-6 IL-1, CXCL14, CCL21, cytokine IL-7, and cytokine IL-7 [3][4][5][6][7][8], and trigger immune cells to react on wound-residing dead cells. The accumulation of fluids in the wounded area promotes the hypoxic condition. Pulmonary hypoxia activates fibroblasts in the blood vessels and leads to vascular remodeling with pulmonary hypertension, and pulmonary hypertension leads to mortality [9] (Figure 1).
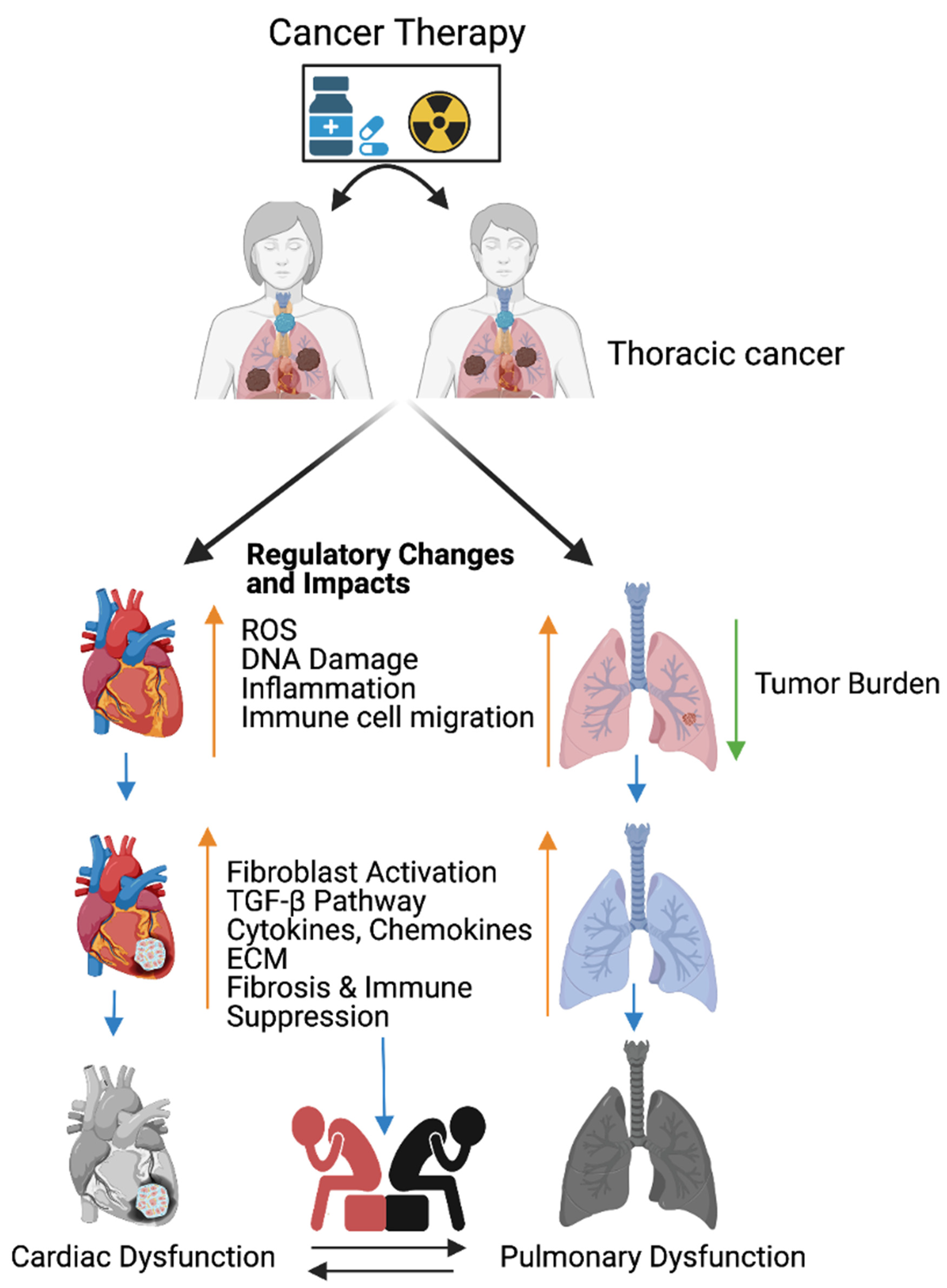
Figure 1. Cancer therapy-induced cardiopulmonary failure. Cartoon illustrates the combination of chemotherapy and ionizing radiation promotes reactive oxygen species (ROS) in thoracic cancer (lung) patients. ROS promotes oxidative stress, DNA damage, immune cell migration, and inflammation, and reduces lung tumor burden. Immune cells at wounded sites secret cytokines, chemokines, and growth factors in cardiopulmonary tissue. Cytokines, chemokines, and growth factors activate myofibroblasts, promote the TGFB pathway, and are involved in the accumulation of the extracellular matrix (ECM). Accumulation of excess ECM induces cardiopulmonary fibrosis, tissue remodeling, immune escape, and cardiopulmonary failure in cancer-free male and female patients who received cancer therapy (chemoradiation therapy). Up arrow indicates the indicated process and the down arrow indicates the reduced tumor burden. This graphic/cartoon is created with BioRender.com agreement # IP22YQRECH.
The combination of chemotherapy and radiation therapy is the most effective treatments for breast, esophageal, and lung cancers. However, the combined therapy promotes early non-symptomatic cardiopulmonary fibrosis responsive risk factors, including the platelet-derived growth factor (PDGF), tumor necrosis factor (TNF), TGFβ, interleukins (IL-1, IL-6, and IL-8), and neutrophil infiltration. Additionally, these treatments induce the late cardiopulmonary fibrogenesis responsive transforming growth factor, IL-4, and IL-13 [10][11]. Histopathological results clearly demonstrate that fibrosis areas are rich in myofibroblast cells, inflammatory cells, and extracellular matrix collagen depositions (ECM) in injured cardiopulmonary organs [12][13]. TGFβ-SMAD-signaling is well studied and characterized in fibrosis models [14][15]. Subsequent accumulation of the ECM in the cardiac tissues leads to myocardial fibrosis, deregulation of heart function, and cardiac failure. Similarly, radiotherapy causes pulmonary pneumonitis and pulmonary fibrosis, which lead to pulmonary remodeling, dysfunction, and pulmonary failure [12] (Figure 1).
2. Activation of M2 Macrophages, Maintenance of Tissue Archistructure, and Extracellular Matrix Remodeling (ECM)
Activated fibroblasts (myofibroblasts) have higher migratory behaviors and produce more ECM components than fibroblasts [16], and the net result is the accumulation of excess ECM in the tissue wounds. Macrophages play important roles in cleaning the excess ECM and maintaining tissue skeletal structure. Macrophages are white blood cells and these cells both migrate into the wounded area or tumor and clear cell debris, while unwanted cancer cells and the extracellular matrix are cleared through the phagocytosis mechanism. Macrophages are grouped under M1 (killer) and M2 (repair) categories and they perform anti-inflammatory roles. Macrophages play a key role in muscle repair as well as in regeneration and maintenance [17][18][19]. M2 macrophages migrate to the site of muscle damage, inflammation, and fibrotic tissue (ECM), and macrophages secrete both matrix metalloproteases (MMPs) and lyse unwanted cells. Additionally, macrophages clear the ECM/scars in the wounded area. Fibrosis is an irreversible process at the wounded site with excess ECM deposition. Matrix metalloproteinases are the enzymes involved in the clearing process of excess ECM, target fibrosis, and maintain the cytoskeletal structure of the tissue. To date, 23 MMPs have been reported and most of which are secreted by macrophages [20]. In addition to MMPs, macrophages secrete pro-inflammatory cytokines and chemokines to attract immune cells neutrophils and natural killer cells to wounded sites to destroy the unwanted cells [21]. Fibroblast proliferation is a key event in the fibrosis, as M2b macrophages inhibit fibroblast proliferation responsive proteins [22], produce MMPs, clear the ECM, and maintain cardiopulmonary cellular homeostasis (Figure 2). Additionally, the ECM is critical for fibrosis and tumor progression, as the ECM blocks the cancer therapeutics and limits the efficacy of cancer drugs. Macrophages are the main source of MMPs and MMPs are capable of destroying the ECM. Currently, developed modified macrophages with the chimeric antigen receptor (CAR) will be an ideal therapeutic to target the ECM and maintain cellular integrity during tissue injury [23].
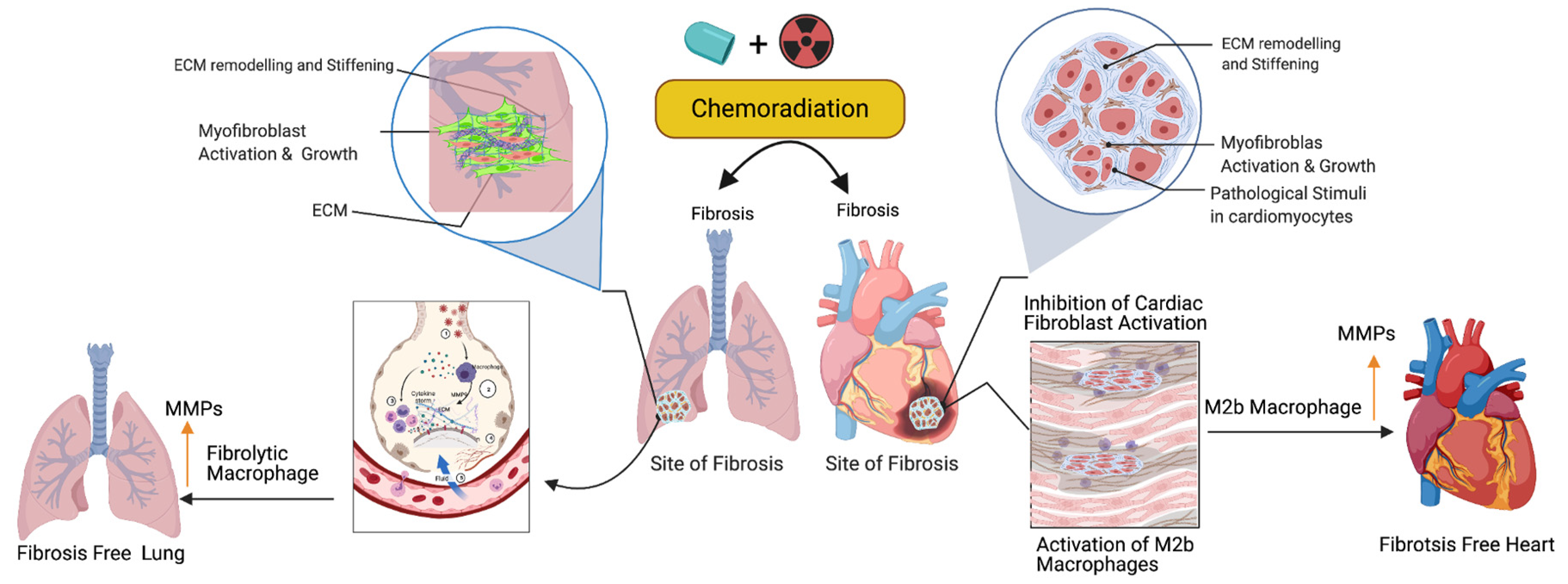
Figure 2. Role of fibrolytic macrophages on the ECM clearance and maintenance of both muscle cell physiology and cellular homeostasis. Following the cancer therapy-induced damage, the immune system triggers the activation of fibrolytic macrophages/M2b macrophages. The activated macrophages migrate to the site of fibrosis and produce or secrete matrix metallopeptidase (MMPs) enzymes to destroy or eliminate excess ECM via phagocytosis/degradation processes, and protect cardiopulmonary muscle cells and their physiological state. Note: Up arrow indicates the upregulation of MMP, M2b macrophages and fibrolytic macrophages. This graphic/cartoon is created with BioRender.com agreement/license # YC22YQS6MB.
3. Biomarker Signatures in Cardiopulmonary Toxicity Following Cancer Therapy
Recent advances in omics technology enabled the identification of molecular changes that underlie the development and progression of human diseases, including cancer. The use of multi-omics, which takes the advantage of technologies such as DNA sequencing, exome sequencing, Chromatin immunoprecipitation (ChIP) sequencing, whole-transcriptome analysis, single-cell transcriptome, 10 X visium genomics analysis, proteomics, and metabolomics, has led to the identification of biomarkers in human diseases, including cancer. Oliver et al., 2019, summarized the importance of the multi-omics approach for the identification of biomarkers in the field of oncology [24]. In the field of oncology, genomics and other omics approaches have identified mechanisms in cancer development, treatment resistance, and the recurrence risk, and these findings have been used in clinical oncology to guide treatment decisions. For example, DNA sequencing and whole-transcriptome sequencing of 100 castration-resistant prostate cancer (CRPC) patient samples identified somatic mutations in TET2, DNAMT3B, and BRAF genes [25]. Interestingly, mapping of the E2F1 cistrome using the transcriptomic approach revealed the role of tumor suppressor RB (retinoblastoma protein) in metabolism and cancer.
Several recent studies have used the state-of-the-art mass spectrometry approach to identify cancer-specific biomarkers in human diseases [26][27] and additional analyses of proteomics data led to the identification of the specific pathways involved in mediating carcinogenesis [28]. Wei Chu et al. identified oral cancer specific biomarkers [29] using mass spectrometry in oral cancer patients’ saliva. [30]. Park et al. employed the metabolomics approach (liquid chromatography coupled with mass spectrometry) and identified four biomarkers including L-octanoylcarnitine, 5-oxyproline, hypoxanthine, and docosahexaenoic acid in breast cancer patients’ plasma. L-octanoylcarnitine serves as an early-stage biomarker with 100% positivity for breast cancer [31]. The application of cutting-edge multi-omics technology, such us whole-exome sequencing, DNA methylation, whole-genome sequencing, metabolomics, protein mass spectrometry, single-cell transcriptomics, and 10 X spatial genomics (visium), is warranted to identify early and late toxicity response biomarkers and cancer relapse genes following cancer therapy in cardiopulmonary tissues.
4. Targeted Therapy-Induced Cardiac Toxicities
One of the recent advancements in cancer therapy is monoclonal antibody-based therapy to target certain molecules to control cancer. Cardiac failure or dysfunction is noted in cancer patients (2–4%) who received anti-PD-1, PD-L1, and bevacizumab (BEV) [32][33], which are monoclonal antibody-based therapies against endothelial cells to target many tumor types; however, this treatment promotes cardiac complications including hypertension, ischemia, and congestive heart failure [34]. The application of the tyrosine kinase inhibitor Erlotinib promotes cardiovascular complications in lung cancer patients [35]. Another monoclonal antibody (necitumumab)-based therapy [36] promotes cardiopulmonary arrest in about 3% of patients. In addition to this, a multi-target tyrosine kinase inhibitor, Nintedanib, targets platelet-derived growth factor (PDGF) receptors and fibroblast growth factors 1, 2, and 3. This agent reduces tumor burden but simultaneously promotes left ventricular dysfunction in cancer patients.
Antioxidants are reductant molecules that react with oxidants and are grouped under two categories, namely endogenous and exogenous. Endogenous antioxidants are produced by the human body, while exogenous antioxidants are produced through the nutrient supply. The exogenous antioxidants fall under the enzymatic and non-enzymatic [37] category. Enzymatic antioxidants possess catalase activity (CAT) that converts hydrogen peroxide to H2O2 and then to water and oxygen. Glutathione (GSH) is considered the most abundant molecule among the endogenous antioxidants. GSH allows to scavenge ROS either directly or indirectly and it directly reacts with O−2 and some other ROS. Aerobic respiration results in increased hydrogen peroxide production, which is detoxified by glutathione peroxide (GPx) by converting two GSH molecules to their oxidized form (GSSG). GSH is recycled by the action of glutathione reductase. Among the antioxidant enzymes, SODs catalyze the dismutation of O2− to H2O2 and catalase stops the formation of OH by converting H2O2 to oxygen and water [38]. Nutrients supply non-enzymatic antioxidants, which reduces the oxidative stress-mediated cardiovascular risk [39]. Additionally, natural extracts (polyphenols) are capable of suppressing ROS and inducing the antioxidant defense mechanism [40][41][42]. Some of the commonly used antioxidants are Vitamin E, vitamin C, polyphenols, non-flavonoids, carotenoids, selenium, lipoic acids, coenzymes Q10 [37]. Earlier studies have shown that CTR increases ROS levels and enhances DNA damage, while CTR-induced cardiopulmonary toxicity is associated with increased ROS levels [43][44][45][46][47][48]. Targeting DNA damage caused by ROS is another promising strategy to reduce chemoradiation side effects. Similar to the prophylactic effect of berberine against rat colon carcinoma, previous studies in lungs demonstrated that ROS scavenging at 20 mg/kg once a day for 6 weeks reduces the inflammation of CTR-induced lung toxicity [49][50]. Furthermore, the systemic administration of Tempol (275 mg/kg) in mice exposed to whole-body irradiation showed the radioprotective effect [51][52][53]. Cisplatin has been used for the treatment of cervical, testicular, esophageal, ovarian, bladder, and lung cancers, and this drug has been associated with cardiopulmonary toxicity [54][55][56][57].
5. Possible Antioxidant Treatments and Prevention of Cancer Therapy-Induced Toxicity
Previous studies have suggested that compounds with antioxidant properties, such as resveratrol [53] and alpha-lipoic acid [58], attenuate cisplatin-induced cardiotoxicities. Antioxidants produced by the human body or consumed from plant products are capable of interacting with and neutralizing ROS, which is induced during oxidative damage/chemoradiation treatment [59][60]. Antioxidants play a key role in controlling cardiopulmonary inflammation and fibrosis [61]. Plant-derived natural antioxidant vitamin A, C, and E from fruits, vegetables, beverages, and cereals [62][63][64][65], and synthetic antioxidant ascorbic acid/vitamin C, glutathione, uric acid, carotenes [66][67][68], and superoxide dismutase (enzyme) are known to suppress oxidative stress [69][70]-induced free radicals (ROS) as well as TGF-β, ERK, and NFκB-mediated signaling, thereby inhibiting fibrogenesis [71] (Figure 3).
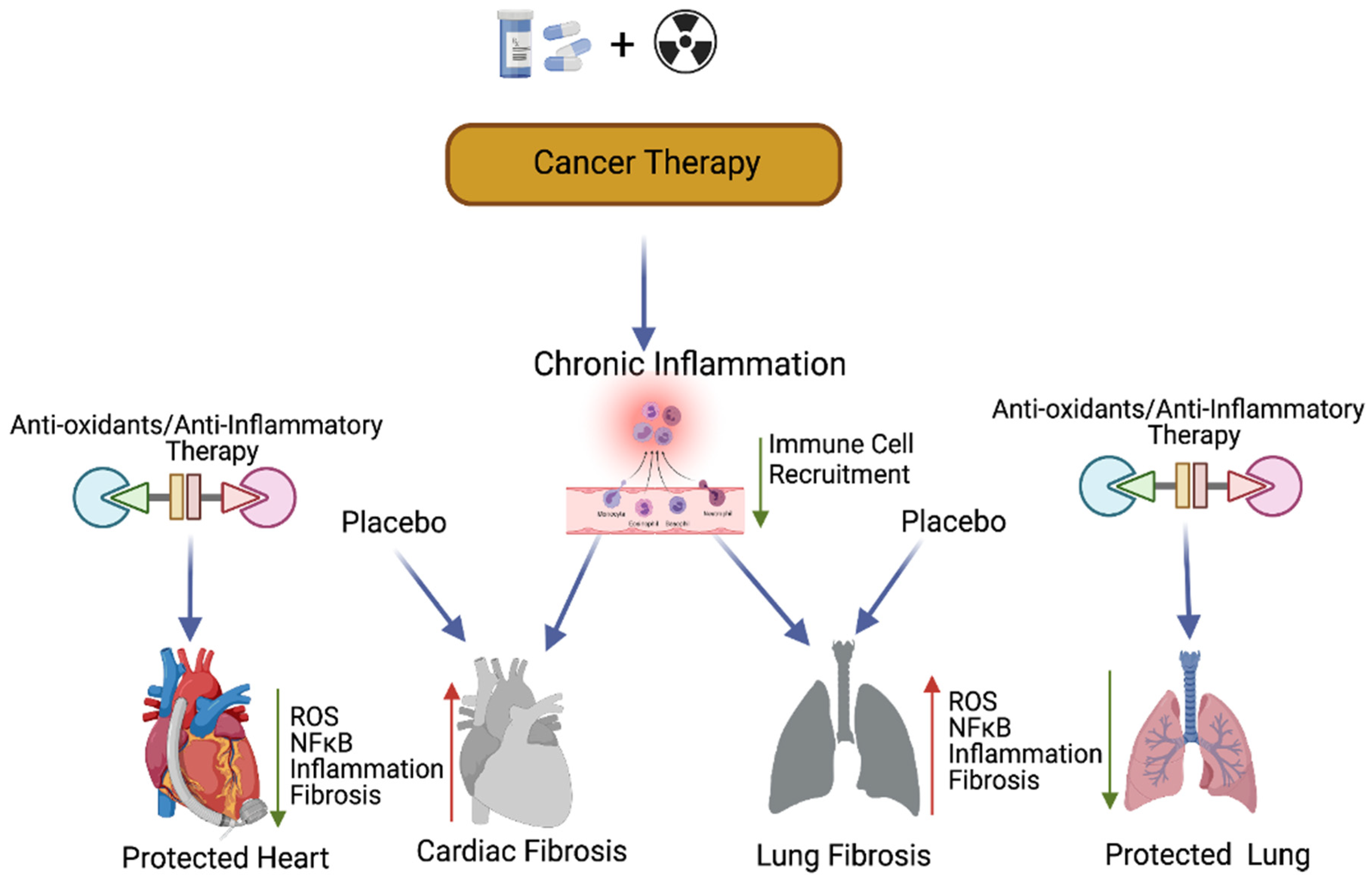
Figure 3. Antioxidants and protection of cancer therapy-induced cardiopulmonary toxicity. This cartoon presentation shows the chemoradiation therapy-promoted cardiopulmonary inflammation, fibrosis via myofibroblast activation, and the NFκB-mediated inflammatory (stress) signaling pathway. The application of antioxidants and inflammatory inhibitors, along with cancer therapy, could protect cardiopulmonary tissue by inhibiting ROS and NFκB-mediated stress signaling. Up arrow indicates the increased levels of ROS, NFκB, inflammation and fibrosis and the down arrow indicates the decreased levels of ROS, NFκB, inflammation, fibrosis and decreased immune cell recruitment. This graphic/cartoon is created with BioRender.com agreement # LD22YQSGUH.
6. Cell Fat Function and Clinical Biomarker Determination
Cancer therapy-induced early cardiopulmonary toxicities are subclinical and not easy to detect in advance. Patients show late symptomatic cancer therapy-induced cardiopulmonary toxicities after 5–10 years post-treatment, which are irreversible. The heart and lung are composed of the heterogenous cell populations. The heart myocardium is composed of 1/3 cardiomyocytes (CMs) and 2/3 remaining fibroblasts, mesothelial, myeloid, lymphoid, adipocytes, pericytes, smooth muscles, endothelial, and neuronal and immune cells [72]. The lung is composed of 40 different cell types including epithelial, smooth muscle, endothelial, nerve, hormone-producing, blood, and structural support cells [73]. Every cell type contributes a specific physiological and pathological function; therefore, it is important to define the cell type specific function when exposed to cancer therapy, which helps to establish clinical biomarkers to target the pathogenesis of fibrosis.
7. Role of Cancer Therapy-Induced Inflammation in Lung Cancer
Chemotherapy is an effective treatment commonly used for various primary tumors and metastatic cancers. In addition to directly destroying cancer cells, chemoradiation has been shown to also induce inflammation with high levels of ROS, IFN-γ, and damage associated proteins (DAMP) [74]. Cardiopulmonary inflammation causes cardiopulmonary tissue damage in response to chemoradiation. In addition, cancer therapy causes genetic alterations (gene mutations due to impaired DNA repair, DNA methylations, and core histone protein modifications) and enhances ROS levels, which leads to chronic inflammation in cardiopulmonary tissues [75][76][77]. Previous studies have demonstrated that radiation exposure leads to senescence and apoptosis of epithelial and endothelial cells, and initiates strong immune response and inflammation in lung tissues [78][79]. Inflammation promotes the epithelial and mesenchymal transition (EMT) in lung cancer cells [80][81]. Previous studies have shown that there were significant changes in the expression of EMT-related proteins in response to cancer therapy in lung cancer cells [82][83]. Many studies indicated that antioxidants reduce ROS levels [84], thereby reducing the inflammation. Anti-inflammatory drugs have been shown to effectively control tumor progression. However, the adverse side effects associated with the different anti-inflammatory drug treatments have limited their full application to cancer therapy [85][86].
Preclinical and clinical studies suggest that there is strong association between chronic inflammation and carcinogenesis [87]. Chronic inflammation is considered to be one of the characteristics of tumor initiation and progression, and chemotherapy-induced chronic inflammation often endows residual cancer cells with resistance and plays a pivot role in promoting therapeutic resistance and cancer progression. The immune system plays major role in maintaining cellular homeostasis, including cell cycle control and tissue remodeling [87]. Diverse populations of leukocytes move to inflammatory areas and produce cytokines, chemokines, and inflammatory metabolites including prostaglandins and leukotrienes.
8. Mechanisms of Inflammation-Induced Carcinogenesis and Cancer Therapy-Induced Tumor Recurrence
Previous studies have implied that chronic inflammation initiates tumorigenesis through DNA damage, excessive replication, and the inhibition of apoptosis and angiogenesis [88]. Inflammation activates Ras, Myc, and p53-signaling, resulting in mitochondrial impairment and increased ROS production [88]. ROS activates NFkB and STAT3-signaling (Figure 4) [89][90], which cause lung carcinogenesis. Anti-inflammatory drugs have been shown to effectively control tumor progression. Recent therapeutic advancements and meta-analyses suggest that the application of non-steroidal anti-inflammatory drugs (NSAID) inhibits inflammation and lung carcinogenesis [91]. However, the adverse side effects associated with the different anti-inflammatory drug treatments in multiple organs including lung, heart, liver, kidney, the digestive stem, and the brain have limited their full application to cancer therapy [91].
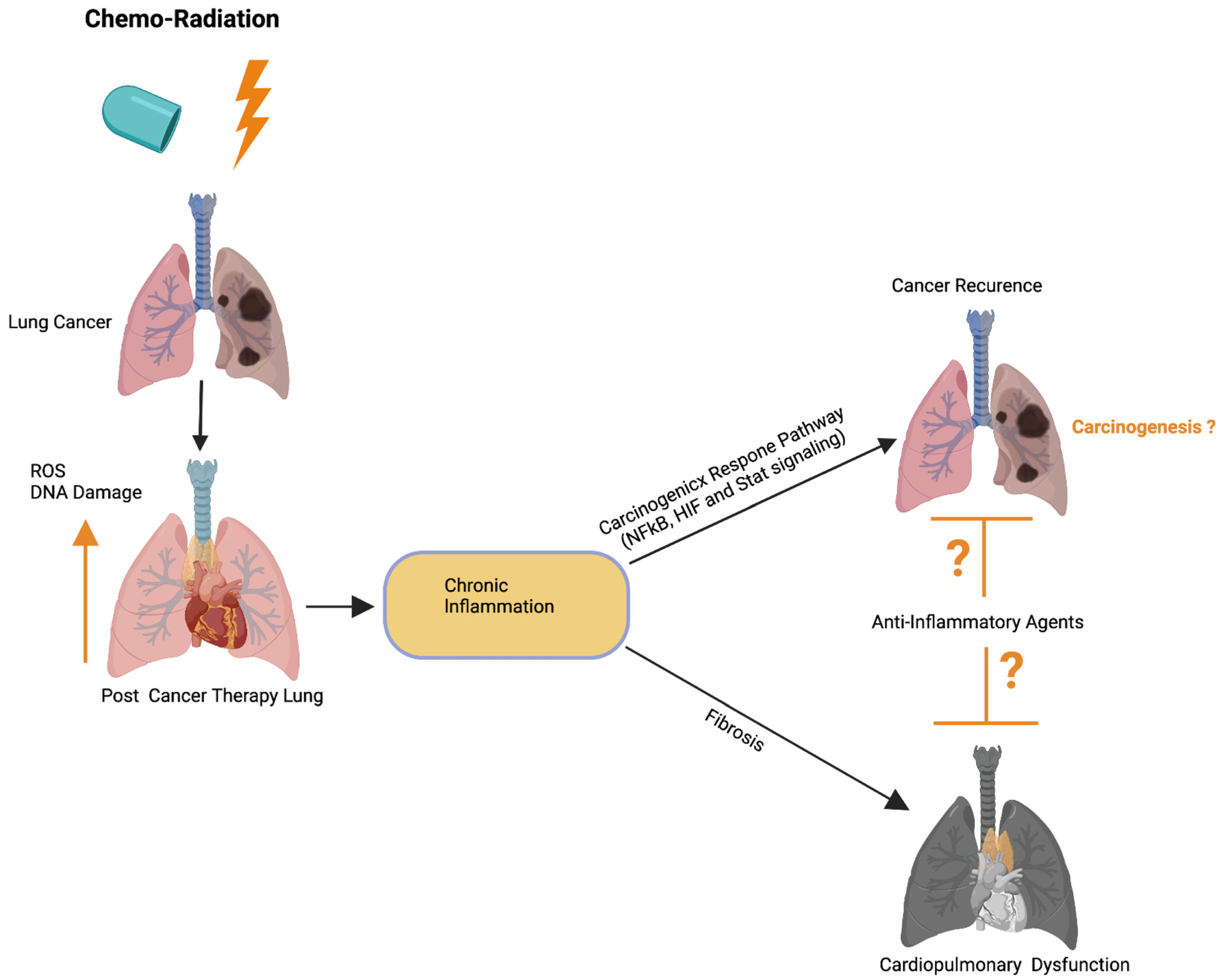
Figure 4. Possible role of cardiopulmonary inflammation on the development of tumor recurrence. Schematics illustrate that chemoradiation therapy inhibits lung tumor burden. Cancer therapy promotes DNA damage and ROS in cardiopulmonary tissue, which leads to chronic cardiopulmonary inflammation. Cardiopulmonary inflammation simultaneously promotes cardiopulmonary fibrosis, organ failure, as well as activation of the carcinogenic response NFkB, HIF, and Stat3 signaling pathways. Possible application of anti-inflammatory therapies could restrict cardiopulmonary fibrosis and tumor recurrence. Up arrow indicates the increased ROS and DNA damage. This graphic/cartoon is created with BioRender.com agreement/license # CU22YQT5TH.
Earlier studies have shown that dying cells (including normal and cancer cells) release growth factors, chemokines, cytokines, interleukins, interferons, and other factors following chemoradiation and these factors prime the carcinogenic-signaling [92]. In addition, dying cells release damage-associated molecular patterns (DAMPs) in response to chemoradiation treatment [93]. DAMPs possibly serve as a ligand for Toll-like receptors (TLRs) expressed on immune cells in the tumor microenvironment (TME) [94]. Cancer therapy not only induces tumor recurrence but also causes drug resistance in patients. Recent study suggests that kinase inhibitors of BRAF, ALK, or EGFR (targeted therapies) promote secretome in melanoma and lung cancer models. RAF and ALK inhibitor-induced secretome increases the proliferation and migration capacity in drug resistance tumor cells [95][96][97][98]. Further studies are warranted to define the mechanism of chemoradiation-induced tumor recurrence and drug resistance in thoracic cancer models.
9. Role of the Inflammatory Microenvironment in Oncogenesis and Metastasis in Lung Cancer
The correlation between inflammation and cancer was revealed based on the fact that cancer originated in sites of chronic inflammation and the tumor biopsies had abundant inflammatory cells [99]. The chronic dysregulated inflammation has been associated with cancer progression in most malignancies [74][100]. Previous studies have implied that the inflammatory tumor microenvironment (TME) is one of the determining factors for the therapeutic efficacy of radiotherapy, chemotherapy, and immunotherapy [101][102]. The inflammatory tumor microenvironment (TME) plays several roles in tumor progression and metastasis. Chronic overexpression of inflammatory mediators in the TME, as seen in patients with lung cancer [103], may lead to increased oncogenesis, progression, invasion, and metastasis. Radiation therapy-induced pro-inflammatory TME in lung cells are possibly involved in activating interconnected networks of cytokines, adhesion molecules, and damage-associated molecular patterns (DAMPs).
Inflammation promotes EMT through its ability to induce the downregulation of epithelial cell-specific proteins and the subsequent upregulation of mesenchymal cell-specific proteins in lung cancer cells [80][81]. This switch from an epithelial to mesenchymal phenotype underscores the importance of the inflammatory microenvironment in the progression of lung cancer. Previous studies have observed that the majority of patients undergoing conventional radiation therapy for locally advanced lung cancer developed relapses and distant metastasis due to resistance [104]. EMT plays a central role in metastasis [105]. Data from previous studies have shown that there was a significant reduction in E-cadherin expression and a remarkable increase in the expression of both α-SMA and vimentin in response to radiation exposure in lung cancer cells [82][83].
This entry is adapted from the peer-reviewed paper 10.3390/ijms221810126
References
- Kang, J.; Jung, M.; Choudhury, M.; Leof, E.B. Transforming growth factor beta induces fibroblasts to express and release the immunomodulatory protein PD-L1 into extracellular vesicles. FASEB J. 2020, 34, 2213–2226.
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503.
- Feghali, C.A.; Wright, T.M. Cytokines in acute and chronic inflammation. Front. Biosci. 1997, 2, d12–d26.
- Gharaee-Kermani, M.; McCullumsmith, R.E.; Charo, I.F.; Kunkel, S.L.; Phan, S.H. CC-chemokine receptor 2 required for bleomycin-induced pulmonary fibrosis. Cytokine 2003, 24, 266–276.
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311.
- Nagineni, C.N.; William, A.; Cherukuri, A.; Samuel, W.; Hooks, J.J.; Detrick, B. Inflammatory cytokines regulate secretion of VEGF and chemokines by human conjunctival fibroblasts: Role in dysfunctional tear syndrome. Cytokine 2016, 78, 16–19.
- Riedel, A.; Shorthouse, D.; Haas, L.; Hall, B.A.; Shields, J. Tumor-induced stromal reprogramming drives lymph node transformation. Nat. Immunol. 2016, 17, 1118–1127.
- Sjöberg, E.; Meyrath, M.; Milde, L.; Herrera, M.; Lövrot, J.; Hägerstrand, D.; Frings, O.; Bartish, M.; Rolny, C.; Sonnhammer, E.; et al. A Novel ACKR2-Dependent Role of Fibroblast-Derived CXCL14 in Epithelial-to-Mesenchymal Transition and Metastasis of Breast Cancer. Clin. Cancer Res. 2019, 25, 3702–3717.
- Lee, P.; Langevitz, P.; Alderdice, C.A.; Aubrey, M.; Baer, P.A.; Baron, M.; Buskila, D.; Dutz, J.P.; Khostanteen, I.; Piper, S. Mortality in systemic sclerosis (scleroderma). Q. J. Med. 1992, 82, 139–148.
- Abid, S.H.; Malhotra, V.; Perry, M.C. Radiation-induced and chemotherapy-induced pulmonary injury. Curr. Opin. Oncol. 2001, 13, 242–248.
- Taunk, N.; Haffty, B.G.; Kostis, J.B.; Egoyal, S. Radiation-Induced Heart Disease: Pathologic Abnormalities and Putative Mechanisms. Front. Oncol. 2015, 5, 39.
- Hanania, A.N.; Mainwaring, W.; Ghebre, Y.T.; Hanania, N.A.; Ludwig, M. Radiation-Induced Lung Injury. Chest 2019, 156, 150–162.
- Madan, R.; Benson, R.; Sharma, D.; Julka, P.; Rath, G. Radiation induced heart disease: Pathogenesis, management and review literature. J. Egypt. Natl. Cancer Inst. 2015, 27, 187–193.
- Camargo, R.D.O.; Abual’Anaz, B.; Rattan, S.G.; Filomeno, K.L.; Dixon, I.M.C. Novel factors that activate and deactivate cardiac fibroblasts: A new perspective for treatment of cardiac fibrosis. Wound Repair Regen. 2021, 29, 667–677.
- Ulmasov, B.; Neuschwander-Tetri, B.A.; Lai, J.; Monastyrskiy, V.; Bhat, T.; Yates, M.P.; Oliva, J.; Prinsen, M.J.; Ruminski, P.G.; Griggs, D.W. Inhibitors of Arg-Gly-Asp-Binding Integrins Reduce Development of Pancreatic Fibrosis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 499–518.
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260.
- Krippendorf, B.B.; Riley, D.A. Distinguishing unloading. Versus reloading-induced changes in rat soleus muscle. Muscle Nerve 1993, 16, 99–108.
- Pierre, B.A.S.; Tidball, J.G. Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J. Appl. Physiol. 1994, 77, 290–297.
- Tidball, J.G.; Berchenko, E.; Frenette, J. Macrophage invasion does not contribute to muscle membrane injury during inflammation. J. Leukoc. Biol. 1999, 65, 492–498.
- Newby, A.C. Metalloproteinase production from macrophages—A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337.
- Patole, P.S.; Schubert, S.; Hildinger, K.; Khandoga, S.; Khandoga, A.; Segerer, S.; Henger, A.; Kretzler, M.; Werner, M.; Krombach, F.; et al. Toll-like receptor-4: Renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int. 2005, 68, 2582–2587.
- Yue, Y.; Huang, S.; Wang, L.; Wu, Z.; Liang, M.; Li, H.; Lv, L.; Li, W.; Wu, Z. M2b Macrophages Regulate Cardiac Fibroblast Activation and Alleviate Cardiac Fibrosis After Reperfusion Injury. Circ. J. 2020, 84, 626–635.
- Zhang, W.; Liu, L.; Su, H.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 2019, 121, 837–845.
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781.
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjöström, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789.
- Brun, A.; Magallanes, M.E.; Del Rio, C.M.; Barrett-Wilt, G.A.; Karasov, W.H.; Caviedes-Vidal, E. A Fast and Accurate Method to Identify and Quantify Enzymes in Brush-Border Membranes: In Situ Hydrolysis Followed by Nano LC-MS/MS. Methods Protoc. 2020, 3, 15.
- MacMullan, M.; Dunn, Z.S.; Graham, N.; Yang, L.; Wang, P. Quantitative Proteomics and Metabolomics Reveal Biomarkers of Disease as Potential Immunotherapy Targets and Indicators of Therapeutic Efficacy. Theranostics 2019, 9, 7872–7888.
- Hristova, V.A.; Chan, D.W. Cancer biomarker discovery and translation: Proteomics and beyond. Expert Rev. Proteom. 2019, 16, 93–103.
- Chu, H.-W.; Chang, K.-P.; Hsu, C.-W.; Chang, I.Y.-F.; Liu, H.-P.; Chen, Y.-T.; Wu, C.-C. Identification of Salivary Biomarkers for Oral Cancer Detection with Untargeted and Targeted Quantitative Proteomics Approaches. Mol. Cell. Proteom. 2019, 18, 1796–1806.
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314.
- Park, J.; Shin, Y.; Kim, T.H.; Kim, D.-H.; Lee, A. Plasma metabolites as possible biomarkers for diagnosis of breast cancer. PLoS ONE 2019, 14, e0225129.
- Sabet, N.S.; Atashbar, S.; Khanlou, E.M.; Kahrizi, F.; Salimi, A. Curcumin attenuates bevacizumab-induced toxicity via suppressing oxidative stress and preventing mitochondrial dysfunction in heart mitochondria. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1447–1457.
- Zaborowska-Szmit, M.; Krzakowski, M.; Kowalski, D.M.; Szmit, S. Cardiovascular Complications of Systemic Therapy in Non-Small-Cell Lung Cancer. J. Clin. Med. 2020, 9, 1268.
- Economopoulou, P.; Kentepozidis, N.; Kotsakis, A.; Kapiris, I. Cancer therapy and cardiovascular risk: Focus on bevacizumab. Cancer Manag. Res. 2015, 7, 133–143.
- Pereira, G.M.; Miller, J.F.; Shevach, E.M. Mechanism of action of cyclosporine A in vivo. II. T cell priming in vivo to alloantigen can be mediated by an IL-2-independent cyclosporine A-resistant pathway. J. Immunol. 1990, 144, 2109–2116.
- Boyiadzis, M.; Foon, K.A. Approved monoclonal antibodies for cancer therapy. Expert Opin. Biol. Ther. 2008, 8, 1151–1158.
- Cammisotto, V.; Nocella, C.; Bartimoccia, S.; Sanguigni, V.; Francomano, D.; Sciarretta, S.; Pastori, D.; Peruzzi, M.; Cavarretta, E.; D’Amico, A.; et al. The Role of Antioxidants Supplementation in Clinical Practice: Focus on Cardiovascular Risk Factors. Antioxidants 2021, 10, 146.
- Lubrano, S.B.V.; Balzan, S. Enzymatic antioxidant system in vascular inflammation and coronary artery disease. World J. Exp. Med. 2015, 5, 218–224.
- Nocella, C.; Cammisotto, V.; Pigozzi, F.; Borrione, P.; Fossati, C.; D’Amico, A.; Cangemi, R.; Peruzzi, M.; Gobbi, G.; Ettorre, E.; et al. Impairment between Oxidant and Antioxidant Systems: Short- and Long-term Implications for Athletes’ Health. Nutrients 2019, 11, 1353.
- Aboul-Enein, H.Y.; Kruk, I.; Kładna, A.; Lichszteld, K.; Michalska, T. Scavenging effects of phenolic compounds on reactive oxygen species. Biopolymers 2007, 86, 222–230.
- Carnevale, R.; Loffredo, L.; Pignatelli, P.; Nocella, C.; Bartimoccia, S.; Di Santo, S.; Martino, F.; Catasca, E.; Perri, L.; Violi, F. Dark chocolate inhibits platelet isoprostanes via NOX2 down-regulation in smokers. J. Thromb. Haemost. 2012, 10, 125–132.
- Li, Y.; Cao, Z.; Zhu, H. Upregulation of endogenous antioxidants and phase 2 enzymes by the red wine polyphenol, resveratrol in cultured aortic smooth muscle cells leads to cytoprotection against oxidative and electrophilic stress. Pharmacol. Res. 2005, 53, 6–15.
- Varga, Z.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Circ. Physiol. 2015, 309, H1453–H1467.
- de Jager, T.L.; Cockrell, A.E.; Du Plessis, S.S. Ultraviolet Light Induced Generation of Reactive Oxygen Species. Adv. Exp. Med. Biol. 2017, 996, 15–23.
- Klebelsberg, D. Psychological characteristics of motorization development. Hefte Unf. 1978, 1978, 348–355.
- Li, Q.; Liang, X.; Yang, Y.; Zeng, X.; Zhong, X.; Huang, C. Panax notoginsengsaponins ameliorate cisplatin-induced mitochondrial injury via the HIF-1α/mitochondria/ROS pathway. FEBS Open Bio 2020, 10, 118–126.
- Meng, X.-M.; Ren, G.-L.; Gao, L.; Yang, Q.; Li, H.-D.; Wu, W.-F.; Huang, C.; Zhang, L.; Lv, X.-W.; Li, J. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab. Investig. 2018, 98, 63–78.
- Vincent, D.T.; Ibrahim, Y.F.; Espey, M.G.; Suzuki, Y.J. The role of antioxidants in the era of cardio-oncology. Cancer Chemother. Pharmacol. 2013, 72, 1157–1168.
- Giuranno, L.; Ient, J.; De Ruysscher, D.; Vooijs, M.A. Radiation-Induced Lung Injury (RILI). Front. Oncol. 2019, 9, 877.
- Ghareeb, A.E.; Moawed, F.S.M.; Ghareeb, D.A.; Kandil, E.I. Potential Prophylactic Effect of Berberine against Rat Colon Carcinoma Induce by 1,2-Dimethyl Hydrazine. Asian Pac. J. Cancer Prev. 2018, 19, 1685–1690.
- Koukourakis, M.I. Radiation damage and radioprotectants: New concepts in the era of molecular medicine. Br. J. Radiol. 2012, 85, 313–330.
- Liebmann, J.; DeLuca, A.M.; Epstein, A.; Steinberg, S.M.; Morstyn, G.; Mitchell, J.B. Protection from Lethal Irradiation by the Combination of Stem Cell Factor and Tempol. Radiat. Res. 1994, 137, 400.
- Wang, J.; He, D.; Zhang, Q.; Han, Y.; Jin, S.; Qi, F. Resveratrol Protects Against Cisplatin-Induced Cardiotoxicity by Alleviating Oxidative Damage. Cancer Biother. Radiopharm. 2009, 24, 675–680.
- Zhang, Z.; Zhang, H. Impact of neoadjuvant chemotherapy and chemoradiotherapy on postoperative cardiopulmonary complications in patients with esophageal cancer. Dis. Esophagus 2017, 30, 1–7.
- Fung, C.; Dinh, P.; Ardeshir-Rouhani-Fard, S.; Schaffer, K.; Fossa, S.D.; Travis, L.B. Toxicities Associated with Cisplatin-Based Chemotherapy and Radiotherapy in Long-Term Testicular Cancer Survivors. Adv. Urol. 2018, 2018, 1–20.
- Kerns, S.L.; Fung, C.; Fossa, S.D.; Dinh, P.C.; Monahan, P.; Sesso, H.D.; Frisina, R.D.; Feldman, D.R.; Hamilton, R.J.; Vaughn, D.; et al. Relationship of Cisplatin-Related Adverse Health Outcomes With Disability and Unemployment Among Testicular Cancer Survivors. JNCI Cancer Spectr. 2020, 4, pkaa022.
- Rabinowits, M.; Souhami, L.; Gil, R.A.; Andrade, C.A.V.; Paiva, H.C. Increased Pulmonary Toxicity with Bleomycin and Cisplatin Chemotherapy Combinations. Am. J. Clin. Oncol. 1990, 13, 132–138.
- El-Awady, E.-S.E.; Moustafa, Y.M.; Abo-Elmatty, D.; Radwan, A. Cisplatin-induced cardiotoxicity: Mechanisms and cardioprotective strategies. Eur. J. Pharmacol. 2011, 650, 335–341.
- Davis, C.D.; Tsuji, P.A.; Milner, J.A. Selenoproteins and Cancer Prevention. Annu. Rev. Nutr. 2012, 32, 73–95.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84.
- Kliment, C.R.; Suliman, H.B.; Tobolewski, J.M.; Reynolds, C.M.; Day, B.J.; Zhu, X.; McTiernan, C.F.; McGaffin, K.R.; Piantadosi, C.A.; Oury, T.D. Extracellular superoxide dismutase regulates cardiac function and fibrosis. J. Mol. Cell. Cardiol. 2009, 47, 730–742.
- Adhami, V.M.; Mukhtar, H. Anti-oxidants from green tea and pomegranate for chemoprevention of prostate cancer. Mol. Biotechnol. 2007, 37, 52–57.
- Mániková, D.; Šestáková, Z.; Rendeková, J.; Vlasáková, D.; Lukáčová, P.; Paegle, E.; Arsenyan, P.; Chovanec, M. Resveratrol-Inspired Benzoselenophenes Act as Anti-Oxidants in Yeast. Molecules 2018, 23, 507.
- Milisav, I.; Ribarič, S.; Poljsak, B. Antioxidant vitamins and ageing. In Subcellular Biochemistry; Springer: New York, NY, USA, 2018; Volume 90, pp. 1–23.
- Tripathi, S.; Kumari, U.; Mazumder, P.M. Ameliorative effects of apple cider vinegar on neurological complications via regulation of oxidative stress markers. J. Food Biochem. 2020, 44, e13504.
- Chakraborthy, A.; Ramani, P.; Sherlin, H.; Premkumar, P.; Natesan, A. Antioxidant and pro-oxidant activity of Vitamin C in oral environment. Indian J. Dent. Res. 2014, 25, 499.
- Njus, D.; Kelley, P.M.; Tu, Y.-J.; Schlegel, H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free. Radic. Biol. Med. 2020, 159, 37–43.
- Sorice, A.; Guerriero, E.; Capone, F.; Colonna, G.; Castello, G.; Costantini, S. Ascorbic Acid: Its Role in Immune System and Chronic Inflammation Diseases. Mini-Rev. Med. Chem. 2014, 14, 444–452.
- Bresciani, G.; da Cruz, I.B.M.; González-Gallego, J. Manganese Superoxide Dismutase and Oxidative Stress Modulation. Adv. Virus Res. 2015, 68, 87–130.
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928.
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40.
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243.
- Franks, T.J.; Colby, T.V.; Travis, W.D.; Tuder, R.M.; Reynolds, H.Y.; Brody, A.R.; Cardoso, W.V.; Crystal, R.G.; Drake, C.J.; Engelhardt, J.; et al. Resident Cellular Components of the Human Lung: Current Knowledge and Goals for Research on Cell Phenotyping and Function. Proc. Am. Thorac. Soc. 2008, 5, 763–766.
- Grivennikov, S.I.; Greten, F.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899.
- Molinaro, C.; Martoriati, A.; Cailliau, K. Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies. Cancers 2021, 13, 3819.
- Antwih, D.A.; Gabbara, K.M.; Lancaster, W.D.; Ruden, D.M.; Zielske, S.P. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics 2013, 8, 839–848.
- Deng, J.-S.; Jiang, W.-P.; Chen, C.-C.; Lee, L.-Y.; Li, P.-Y.; Huang, W.-C.; Liao, J.-C.; Chen, H.-Y.; Huang, S.-S.; Huang, G.-J. Cordyceps cicadae Mycelia Ameliorate Cisplatin-Induced Acute Kidney Injury by Suppressing the TLR4/NF-κB/MAPK and Activating the HO-1/Nrf2 and Sirt-1/AMPK Pathways in Mice. Oxid. Med. Cell. Longev. 2020, 2020, 1–17.
- Citrin, D.E.; Prasanna, P.G.S.; Walker, A.J.; Freeman, M.L.; Eke, I.; Barcellos-Hoff, M.H.; Arankalayil, M.J.; Cohen, E.P.; Wilkins, R.; Ahmed, M.M.; et al. Radiation-Induced Fibrosis: Mechanisms and Opportunities to Mitigate. Report of an NCI Workshop, September 19, 2016. Radiat. Res. 2017, 188, 1–20.
- Johnston, C.J.; Hernady, E.; Reed, C.; Thurston, S.W.; Finkelstein, J.N.; Williams, J.P. Early Alterations in Cytokine Expression in Adult Compared to Developing Lung in Mice after Radiation Exposure. Radiat. Res. 2010, 173, 522–535.
- Heinrich, E.L.; Walser, T.C.; Krysan, K.; Liclican, E.L.; Grant, J.L.; Rodriguez, N.L.; Dubinett, S.M. The Inflammatory Tumor Microenvironment, Epithelial Mesenchymal Transition and Lung Carcinogenesis. Cancer Microenviron. 2012, 5, 5–18.
- Willis, B.C.; Borok, Z. TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L525–L534.
- Liu, W.; Huang, Y.-J.; Liu, C.; Yang, Y.-Y.; Liu, H.; Cui, J.-G.; Cheng, Y.; Gao, F.; Cai, J.-M.; Li, B.-L. Inhibition of TBK1 attenuates radiation-induced epithelial–mesenchymal transition of A549 human lung cancer cells via activation of GSK-3β and repression of ZEB1. Lab. Investig. 2014, 94, 362–370.
- Lu, J.; Zhong, Y.; Chen, J.; Lin, X.; Lin, Z.; Wang, N.; Lin, S. Radiation Enhances the Epithelial– Mesenchymal Transition of A549 Cells via miR3591-5p/USP33/PPM1A. Cell. Physiol. Biochem. 2018, 50, 721–733.
- Dennis, J.M.; Witting, P.K. Protective role for antioxidants in acute kidney disease. Nutrients 2017, 9, 718.
- Bonifant, C.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011.
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25.
- Gomes, M.; Teixeira, A.L.; Coelho, A.; Araújo, A.; Medeiros, R. The Role of Inflammation in Lung Cancer. Adv. Exp. Med. Biol. 2014, 816, 1–23.
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology 2011, 25, 400.
- Multhoff, G.; Molls, M.; Radons, J. Chronic Inflammation in Cancer Development. Front. Immunol. 2012, 2, 98.
- O’Callaghan, D.S.; O’Donnell, D.; O’Connell, F.; O’Byrne, K.J. The Role of Inflammation in the Pathogenesis of Non-small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 2024–2036.
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147.
- Jiang, M.; Gu, D.-N.; Dai, J.-J.; Huang, Q.; Tian, L. Dark Side of Cytotoxic Therapy: Chemoradiation-Induced Cell Death and Tumor Repopulation. Trends Cancer 2020, 6, 419–431.
- Patidar, A.; Selvaraj, S.; Sarode, A.; Chauhan, P.; Chattopadhyay, D.; Saha, B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine 2018, 104, 114–123.
- Fang, H.; Ang, B.; Xu, X.; Huang, X.; Wu, Y.; Sun, Y.; Wang, W.; Li, N.; Cao, X.; Wan, T. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell. Mol. Immunol. 2014, 11, 150–159.
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758.
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell 2012, 22, 668–682.
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409.
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372.
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545.
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867.
- Crusz, S.M.; Balkwill, F. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596.
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309.
- Strang, R.R. Preliminary report on a new anti-Parkinson drug, UCB 1549. Acta Neurol. Psychiatr. Belg. 1966, 66, 771–776.
- Blackhall, F.; Faivre-Finn, C. Treatment of limited small cell lung cancer: An old or new challenge? Curr. Opin. Oncol. 2011, 23, 158–162.
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39.
This entry is offline, you can click here to edit this entry!