Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
PTPN2 (protein tyrosine phosphatase non-receptor 2), also called TCPTP (T cell protein tyrosine phosphatase), is a member of the PTP family signaling proteins. Phosphotyrosine-based signaling of this non-transmembrane protein is essential for regulating cell growth, development, differentiation, survival, and migration.
- PTPN2
- inflammation
- immunology
1. Anti-Inflammatory Role of PTPN2
Abnormal expression of PTPN2 results in the occurrence of many inflammatory diseases. The anti-inflammatory role of PTPN2 is highlighted by the fact that PTPN2-deficient mice die a few weeks after birth because of systemic inflammation and severe colitis [22]. Loss of functional variants in PTPN2 is associated with an increased risk of developing chronic inflammatory disorders [12].
2. PTPN2 in Intestinal Inflammation
The PTPN2 gene has gained clinical interest recently since many SNPs in the 18p11 locus are associated with chronic inflammatory diseases [23]. More specifically, recent studies have demonstrated that the rs2542151 SNP is associated with inflammatory bowel disease, Crohn’s disease, and ulcerative colitis [24]. At the RNA and protein levels, PTPN2 mRNA and protein expression levels are elevated within the epithelial cells in inflammatory bowel disease (IBD). In addition, IBD-related inflammatory cytokines (e.g., IFN-γ, TNF) increase PTPN2 expression in an intestinal epithelial cell (IEC) line [25]. The deficiency of PTPN2 exacerbates barrier dysfunction after IFN-γ treatment [26]. Thus, IBD-inflammatory cytokines demonstrate a negative feedback loop whereby the loop triggers the expression of its own negative regulator of the signaling pathway [25].
PTPN2 influences intestinal inflammation by regulating (1) intestinal barrier function, (2) inflammatory factors, and/or (3) associated inflammatory immune cells.
The lack of PTPN2 expression in IECs significantly impacts the formation of an effective intestinal barrier. PTPN2 knockdown in IECs exacerbates the barrier disorders caused by IFN-γ treatment [26]. In addition, PTPN2 knockdown in IECs increases the expression of cation-selective pore-forming proteins, allowing the paracellular passage of cations into the intestinal lumen, which leads to intestinal fluid loss [26,27]. In addition to the damaged intestinal barrier, the secretion and related signaling mechanisms of inflammatory factors regulated by PTPN2 also affect the inflammatory response in the intestine. PTPN2 deficiency leads to an IFN-γ-mediated barrier defect in chronic intestinal inflammatory diseases associated with STAT1 signaling [26,28]. It also enhances the inhibitory effect of epidermal growth factor on intestinal epithelial chloride secretion [29] and promotes TNFα-induced secretion of cytokines [30]. Additionally, the loss of PTPN2 is associated with TNFα-induced extracellular signal-regulated kinase 1/2 (ERK1/2) and p38, without affecting c-Jun N-terminal kinase (JNK) or NF-κB phosphorylation signaling. In addition, the loss of PTPN2 potentiates TNFα-induced secretion of interleukin 6 (IL-6) and IL-8. These data indicate that PTPN2 activity plays a crucial role in the establishment of chronic inflammatory conditions in the intestine [30]. Moreover, PTPN2 initiates and orchestrates efficient immune responses against bacteria that penetrate the epithelial barrier [31]. The immune cells maintain intestinal homeostasis by removing invading bacteria and dying cells, secreting anti-inflammatory cytokines, and inducing/maintaining tolerance toward commensal bacteria and food particles. Loss of PTPN2 increases the inflammasome activity of macrophages by elevating the phosphorylation of ASC, the essential inflammasome adaptor protein, and leads to elevated IL-1β production. Thus, the loss of PTPN2 in macrophages causes more severe colitis, which may be mitigated by inhibiting IL-1β [22]. In addition, spermidine reduces inflammation by raising the expression and activity of PTPN2 in human THP-1 monocytes, which results in a reduction of STATs and p38 MAPK signaling, and IFN-γ induced expression/secretion of certain pro-inflammatory cytokines [32].
3. PTPN2 in Other Inflammatory Reactions
Atherosclerosis is the primary cause of cardiovascular disease. Systemic inflammation is an important characteristic of atherosclerosis, which is aggravated by the inflammatory factors secreted by pro-inflammatory macrophages. PTPN2 assists in inhibiting the release of inflammatory factors in macrophages via de-phosphorylating p65/p38/STAT3 in an atherosclerosis model. The results indicate that PTPN2 plays a negative role in the occurrence of atherosclerosis by inhibiting the secretion of inflammatory factors in macrophages and may be a treatment candidate for atherosclerosis (Figure 1) [33].
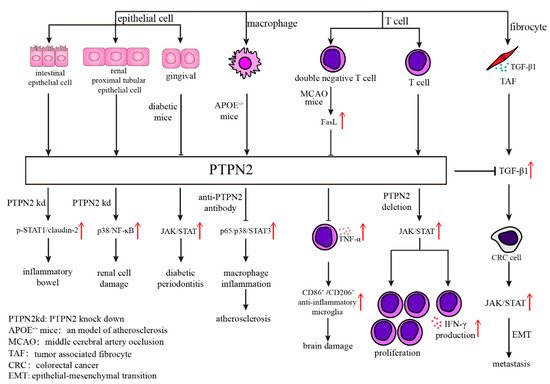
Figure 1. Various mechanisms of PTPN2 in different kinds of cells. Abnormal expression of PTPN2 in epithelial cells will result in many diseases through different mechanisms, such as p-STAT1/claudin-2 in inflammatory bowel, p38/NF-κB in renal cell damage, and JAK/STAT in diabetic periodontitis. The antibody of PTPN2 in APOE−/− mice inhibits atherosclerosis through diminishing p65/p38/STAT3 signaling pathway. What’s more, PTPN2 influences T cell function, including cell proliferation and IFN-γ production, through JAK/STAT signaling pathway.
Neuro-vascular inflammation is characterized by the breakdown of the blood-brain barrier (BBB) and increased endothelial permeability, which leads to cerebral edema, a condition that can occur in a range of illnesses such as stroke, trauma, tumor, infection, and degenerative diseases [34,35,36,37]. Angiopoietin-1 (Ang-1) diminishes thrombin-induced breakdown of the BBB by mediating disruption of tight junctions (TJs), which are involved in tyrosine phosphorylation of Occludin (a major component of TJs). Depletion of PTPN2 eliminates Ang-1 function that promotes tyrosine de-phosphorylation of Occludin, and endothelial hyperpermeability. The results indicate that PTPN2 blockage via mediating tyrosine phosphorylation of Occludin is closely tied to maintaining BBB function and may be a novel therapeutic target for neuro-inflammatory disorders associated with BBB disruption [38]. Regarding neuroinflammation after ischemic stroke, CD3+CD4−CD8− T cells (double-negative T cells; DNTs) dramatically increased in stroke patients and in a mouse model in a time-dependent manner, which exacerbates cerebral immune and inflammatory responses and ischemic brain injury via TNF-α production and the following proinflammatory microglial activation. This process involves the FasL/PTPN2/TNF-α signaling pathway, in which FasL activation promotes TNF-α production in DNTs, where PTPN2 serves as a negative regulator of FasL signaling to suppress TNF-α secretion (Figure 2) [39].
PTPN2 also contributes to the regulation of renal inflammation. In HK-2 cells (a classical model of sepsis-induced renal injury in vitro), PTPN2 reduced LPS-induced inflammatory cytokine release and cell death via modulating p38 MAPK/NF-κB signaling, which alleviated renal cell damage by playing a nephron-protective role in sepsis-induced renal injury (Figure 2) [40].
Moreover, PTPN2 plays a part in inflammation-associated metabolic disorders, such as diabetes-related diseases. Diabetic nephropathy (DN) is a chronic inflammatory kidney disease caused by diabetes. Recent studies have shown that PTPN2 exerts protective effects by ameliorating metabolic disorders and suppressing micro-inflammation via the STAT signaling pathway, which suggests PTPN2 is a potential target for the treatment of human DN [41]. In addition, the interaction between PTPN2 and JAK/STAT pathway may contribute to the development of diabetic periodontitis [42].
4. PTPN2 Regulates the Development and Redistribution of T Lymphocytes
Both the development of T lymphocytes in the thymus and the activation of mature T lymphocytes in secondary lymphoid tissues require that the lymphocytes respond adaptively to environmental signaling molecules. The T cell receptor (TCR) interaction with the MHC/antigen peptide complex, together with the CD4 and CD8 co-receptors’ interaction with the co-stimulatory molecules and cytokine receptor-mediated signals, activates the TCR signaling pathway and leads to an immune response. The lymphocyte-specific protein tyrosine kinase (Lck) and proto-oncogene tyrosine-protein kinase (Fyn) kinases, members of the Src family of non-receptor tyrosine kinases, influence T lymphocyte activation, differentiation, and tolerance [43]. Lck and Fyn are proximal signal proteins that are activated in the TCR signaling pathway.
Protein tyrosine phosphatases (PTPs) play an important role in T lymphocyte development and function. Among these, PTPN2 has different qualitative or quantitative effects on the early activation, proliferation, survival of mature T lymphocytes, differentiation of T cells, and regulation of T lymphocyte subsets.
T cell progenitors transition through T cell differentiation through the IL-7-STAT5 axis, of which the target genes change dynamically. IL-7R modulates gene expression via the JAK-STAT pathways, and STAT5 is the primary STAT family member activated downstream of IL-7R [44]. PTPN2, a negative regulator of IL-7R-STAT signaling, contributes to the nature of STAT-mediated gene targeting in T cell differentiation. A lack of PTPN2 expression results in an abnormal interferon-response gene profile due to amplified phosphorylation of STAT family members, which leads to the deregulation of early development checkpoints and the ensuing inefficient differentiation of CD4+ CD8+ double-positive lymphocytes [45]. PTPN2 also dephosphorylates TCR-proximal kinases, such as Lck and Fyn, which results in the increase of the threshold for T cell activation, and subsequent reduction of the sensitivity to low-affinity antigens during T cell signaling [46,47,48] Generally speaking, lack of PTPN2 causes broad changes in the expression and phosphorylation of T cell expansion and survival-associated proteins, which renders cells less dependent on survival-promoting cytokines. Thus, PTPN2 deficiency leads to: (1) augment of programmed T cell expansion and survival capacity of activated T cells [11]; (2) enhancement of T cell signaling [48]; (3) promotion of CD8+ T cell responses after antigen cross-presentation [46]. In lymphopenia-induced proliferation (LIP), a condition that contributes to the onset of inflammatory bowel disease, rheumatoid arthritis, and type I diabetes [49], some T cells expand due to the recognition of self-antigens and/or cytokines, particularly IL-7. PTPN2 is strongly engaged in this process, with its expression elevated in naive T cells that leave the thymus to restrict homoeostatic T cell proliferation and prevent excess responses to self-antigens in the periphery [47]. Consequently, PTPN2 deficiency leads to an elevated T cell receptor-dependent response and the further development of autoimmunity. As a result, negative regulation by PTPN2 in T cells plays an important role in preventing the development of autoimmune and inflammatory disorders.
Moreover, PTPN2 engages in the regulation of T lymphocyte subsets. The PTPN2rs1893217(C) risk allele with reduced PTPN2 expression causes decreased IL-2R/pSTAT5 signaling and further reduces FOXP3 expression in activated CD4+ T cells. This leads to a decrease in CD4+ FOXP3+ T cells [50]. In addition, PTPN2 deletion in CD8+ T lymphocytes increases the production, proliferation, and cytotoxicity of a Tim-3+ terminally exhausted subset without altering the number of a Slamf6+ progenitor exhausted subset in lymphocytic choriomeningitis virus clone 13 infection [51]. Additionally, a lack of Ptpn2 expression in CD8+ T cells leads to a reduction in tissue-resident memory T cells and the proportion of memory precursor cells [52]. Although PTPN2 was originally cloned from a human T cell cDNA library, it also participates in the regulation of other immune cells. Dendritic cells (DCs), which hold a crucial position between innate and adaptive immunity, modulate immunological tolerance and immune responses. Thus, DCs play a significant role in tissue homeostasis and the prevention of autoimmune responses [53]. The lack of PTPN2 in mouse DCs altered the proportion of myeloid and lymphoid immune cells in the skin, liver, lung, and kidney [54]. In THP-1 cells, loss of PTPN2 participates in inflammation-related events by promoting IFN-γ-induced STAT signaling, and IL-6 or MCP-1 secretion [55].
On the whole, PTPN2, as a prominent regulator of inflammatory and immune/autoimmune response, is a potential target to manage in order to maintain tissue tolerance.
5. The Role of PTPN2 in Immune Cells
Since PTPN2 influences the production, differentiation, and distribution of immune cells, researchers have turned the spotlight to the role of PTPN2 in the anti-tumor immune response.
Tumors evade the cytotoxicity of the immune response primarily by (1) avoiding immune recognition and (2) instigating an immunosuppressive TME. On the one hand, PTPN2 participates in tumor avoidance of immune recognition. PTPN2 deficiency boosts the expression of human leukocyte antigens (HLAs), which causes a reduction of immune escape by presenting more antigens [13]. Additionally, PTPN2 deficiency promotes the production and secretion of T cell effector molecules, such as TNF-α and INF-γ, which increases the likelihood of detection by T cells [56].
On the other hand, PTPN2 is involved in the induction of immunosuppressive TME. PTPN2 deletion in T cells increases proliferation through elevation of JAK/STAT signaling and INF-γ production [57,58], which encourages CD4+ Th1 cell development and activation, as well as enhanced CD8+ T cell cytotoxicity [59]. PTPN2 deletion in CD8+ T cells also boosts the generation, proliferation, and cytotoxicity of Tim-3+ terminally exhausted subpopulation without altering the Slamf6+ progenitor exhausted subpopulation, which enhances anti-tumor responses and improves tumor control [51]. Additionally, PTPN2 deletion in T cells enhances the efficacy of anti-PD-1 therapy and achieves complete tumor clearance in a murine colorectal cancer model [13,59]. PTPN2 deletion of the immune system also resulted in MC38 tumor clearance and improved PD-1 checkpoint blockade responses to B16 tumors [51]. The above findings indicate that PTPN2 deletion sensitizes cancer cells to immune checkpoint therapy.
Chimeric antigen receptor-T (CAR-T) cell therapy plays a prominent role in cancer treatment. The deletion of PTPN2 in HER-2-specific CAR-T cells activates Src family kinase LCK and STAT5 signaling, enabling CAR-T cells to be activated and homed in CXCL9/10-expressing tumors to eliminate HER-2+ breast tumors in vivo. These findings define PTPN2 as a promising target for enhancing T cell tumor infiltration and tumor cytotoxicity [56].
Moreover, PTPN2 exerts a tumor-associated immunity function in other immune cells. For example, PTPN2 deficiency consistently enhances the cytotoxicity of NK cells [60]. PTPN2 deficiency in macrophages also induces the formation of inflammasomes, which convert pro-IL-1ß and pro-IL18 into their active forms via protease caspase-1 cleavage. IL-1ß has powerful pro-inflammatory properties, whereas IL-18 promotes the induction of IFN-γ-expressing cells, recruitment of pro-inflammatory phagocytes, and differentiation of Th17 lymphocytes, Th1 lymphocytes, CD8+ cytotoxic T lymphocytes, and NK cells, all of which contribute to anti-cancer immunity [61].
This entry is adapted from the peer-reviewed paper 10.3390/ijms231710025
This entry is offline, you can click here to edit this entry!