Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Pemphigus vulgaris (PV) is an IgG autoantibody-mediated, potentially fatal mucocutaneous disease manifested by progressive non-healing erosions and blisters. Beyond acting to inhibit adhesion molecules, PVIgGs elicit a unique process of programmed cell death and detachment of epidermal keratinocytes termed apoptolysis. Mitochondrial damage by antimitochondrial antibodies (AMA) has proven to be a critical link in this process.
- pemphigus vulgaris
- apoptosis
- antimitochondrial autoantibodies
1. Introduction
Pemphigus encompasses a family of rare, potentially lethal autoimmune blistering dermatoses involving the skin and mucosal surfaces. The word ‘pemphigus’ derives from the Greek “pemphix”, which means blister. Its earliest use dates back to Hippocrates in 460–370 B.C. [1]. In modern history, the disease was first described by an Irish physician in 1788 [2], however, the current understanding of pemphigus pathophysiology began in 1964 with the discovery of autoantibodies in the sera of pemphigus vulgaris (PV) patients directed against the cell surface of keratinocytes [3]. The disease is associated with both circulating and tissue-bound IgG autoantibodies, and manifested by the loss of cell–cell adhesion of keratinocytes (acantholysis), and formation of non-healing suprabasal intraepidermal blisters.
IgG antibodies against desmoglein-1 (Dsg1) and desmoglein-3 (Dsg3), calcium-dependent cell adhesion molecules of the cadherin family, have been considered to play a primary role in the development of PV. However, explanation of the pathogenesis remains controversial [4]. Clinically, the detection of anti-Dsg3 reactivity, with or without anti-Dsg1 reactivity, is helpful in diagnosing PV. However, there have been a number of reports of patients in whom no reactivity was found, challenging the notion of an exclusive role these proteins have in the biologic mechanism of keratinocyte cohesion and their autoantibodies in blister formation in patients (reviewed in Ref. [5]).
Proteomic studies have led to the discovery of additional major defined types of non-Dsg proteins targeted by pemphigus autoantibodies, including: mitochondrial proteins, desmocollin 1 and 3 (Dsc1 and Dsc3), various nicotinic and muscarinic acetylcholine receptor subtypes, thyroid peroxidase, human leukocyte antigen (HLA) molecules, and secretory pathway Ca2+/Mn2+-ATPase isoform 1 (SPCA1) encoded by the ATP2C1 gene, which is mutated in Hailey-Hailey disease [6]. A “multiple hit” hypothesis has been proposed [7], wherein various non-Dsg autoantibodies against keratinocytes act synergistically with anti-Dsg autoantibodies to cause blistering. These non-Dsg autoantibodies can induce changes seen in PV, including keratinocyte shrinkage, cell–cell detachment, and triggering of apoptotic signaling events (reviewed in Ref. [4]). For example, non-Dsg autoantibodies against Dsc3, M3 muscarinic acetylcholine receptor (M3AR), and SPCA1 isolated from the sera of patients with anti-Dsg1/3 autoantibody-negative PV were found to be pathogenic, working synergistically with each other to cause acantholysis [8]. Thus, recent discoveries of numerous non-Dsg autoantibody species further develop the understanding of PV and implicate additional cell metabolism and signaling pathways involved in acantholysis [6][9].
2. Apoptolysis
Beyond acting at the keratinocyte cell membrane to block the function of adhesion molecules, PVIgGs elicit the signaling events that trigger the keratinocyte cell death program. The term “apoptolysis” has been coined to describe the distinct autoantibody-induced process of keratinocyte structural damage and detachment (acantholysis) followed by death (apoptosis), which is unique to PV. Acantholysis and apoptosis are inseparable in PV and are mediated by the same cell death enzymes [10]. Apoptosis refers to programmed cell death—a pathway not activated by inflammation, but rather by cysteine aspartate proteases, or caspases. This cell death pathway can be activated by cellular damage (intrinsic pathway) or by signaling molecules (extrinsic, death receptor-initiated pathway), and ultimately results in the formation of apoptotic bodies which are then cleared by phagocytic cells [11]. The best-known cell death pathways are apoptosis, oncosis, and necrosis, however others have recently been described by the Nomenclature Committee on Cell Death (NCCD) [12]. Additional, newly described apoptolytic pathways that play a role in the pathophysiology of skin blistering characteristic of PV should be further investigated.
Mitochondria play a critical role in programmed cell death (reviewed in Ref. [13]). Initiation of apoptotic pathways ultimately disrupt the inner mitochondrial membrane, resulting in loss of the mitochondrial transmembrane potential and leakage of pro-apoptotic proteins into the cell cytosol, including the release of cytochrome c (CytC), a marker for mitochondrial outer membrane permealization and early apoptosis, and subsequent activation of caspases [14]. The disruption of mitochondrial energy production, combined with cleavage of adhesion and structural molecules, causes cytoskeleton collapse and the keratinocyte to shrink [10]. The fundamental feature of apoptolysis is that anti-keratinocyte antibodies in PV cause basal keratinocytes only to shrink, but not to die, giving rise to their “tombstone” appearance on histopathology. This is distinct from the classic apoptotic processes in the epidermis of patients with Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis, in which apoptosis leads to sloughing of the entire epidermis, including its basal layer [15].
In PV, the apoptotic pathway is activated long before morphological evidence of acantholysis [16][17]. The hypothetical sequence of apoptolysis development in PV has five consecutive steps: (i) Pathogenic autoantibodies bind PV antigens on the surface of keratinocytes and pro-apoptolytic signals are transduced. (ii) Activation of EGFR, mTOR, Src, p38 MAPK and other signaling pathways increase intracellular calcium and initiate programmed cell death enzymatic cascades predominately in basal keratinocytes. (iii) Executioner caspases cleave tonofilaments, leading to their collapse and retraction, while inter-desmosomal adhesion complexes are phosphorylated and dissociate. This results in basal cell contraction, a crossing step of both the apoptotic and early acantholytic pathways. The majority of desmosomes remain intact and bridge collapsing keratinocytes. (iv) The continued degradation of structural proteins by the programmed cell death enzymes lead to cytoskeleton collapse and complete separation of shrinking keratinocytes (visible acantholysis). The sloughed cell membrane pieces trigger production of scavenging (secondary) autoantibodies to Dsg, Dsc, E-cadherin and other adhesion molecules attached to the cell membrane. (v) The end result is rounding up and apoptotic death of acantholytic cells resulting from irreversible damage to mitochondrial and nuclear proteins by the same cell death enzymes giving rise to a “tombstone” appearance of the surviving basal keratinocytes [10]. The more recent observations of the pathogenic role of AMA, however, indicate that damage of mitochondria occurs at an early stage of apoptolysis.
3. Mitochondrial Damage by AMA in PV
Patients with PV produce PVIgG antibodies targeting a variety of proteins, including those at the inner and outer mitochondrial membrane, as well as the mitochondrial matrix [18][19][20]. Utilizing protein microarray, the most common antigen targets recognized by AMA in PV have been identified from a large cohort of patients (Table 1) [18]. Based on the known functions of these proteins, AMA likely lead to mitochondrial dysfunction by altering cellular ability to produce or inactivate reactive oxygen species, perform oxidative phosphorylation, and participate in oxygen respiration. The exact mechanism of mitochondrial damage likely varies greatly among PV patients, consistent with the strikingly wide spectrum of disease severity and treatment response [20]. Importantly, absorption of AMA inhibits the ability of PVIgGs to induce keratinocyte detachment and blistering [19].
Table 1. Mitochondrial autoantibodies in patients with PV (adapted from Kalantari et al. [18]) *.
| Symbol | Antigen | Localization on Mitochondria | Frequency | |
|---|---|---|---|---|
| PV (%) | Control (%) | |||
| ABAT-V1 | 4-Aminobutyrate aminotransferase, mitochondrial; 50 kDa | Matrix | 19 | 4 |
| ALDH4A1 | Aldehyde dehydrogenase 4 family, member A1 | Matrix | 23 | 5 |
| CPT1B | Carnitine O-palmitoyltransferase 1B | Outer membrane | 18 | 5 |
| CRAT | Carnitine O-acetyltransferase | Inner membrane | 28 | 7 |
| CYB5B | Cytochrome b5 type B; 21 kDa | Outer membrane | 19 | 1 |
| ETFA | Electron transfer flavoprotein, α protein | Matrix | 19 | 4 |
| ETFB | Electron transfer flavoprotein, β protein | Matrix | 21 | 3 |
| FDXR-V2 | NADPH:adrenodoxin oxidoreductase | Matrix | 25 | 6 |
| FH | Fumarate hydratase (fumarase) | Mitochondrion | 29 | 3 |
| MAOB | Amine oxidase (flavin-containing) B | Outer membrane | 27 | 5 |
| ME2 | NAD-dependent malic enzyme | Matrix | 18 | 6 |
| ME3 | NADP-dependent malic enzyme, mitochondrial | Matrix | 23 | 8 |
| MLYCD | Malonyl-CoA decarboxylase | Mitochondrion | 29 | 4 |
| NDUFA9 | NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 9; 39 kDa | Matrix | 20 | 3 |
| NDUFA13 | NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 13; 16 kDa | Inner membrane | 24 | 6 |
| NDUFB10 | NADH dehydrogenase [ubiquinone] 1β subcomplex subunit 10 | Matrix | 17 | 2 |
| NDUFV3 | NADH dehydrogenase [ubiquinone] flavoprotein 3; 9 kDa | Inner membrane | 19 | 4 |
| NDUFS6 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 6; 13 kDa | Inner membrane | 24 | 6 |
| PC | Pyruvate carboxylase | Matrix | 32 | 5 |
| PDK4 | Pyruvate dehydrogenase kinase, isozyme 4 | Matrix | 24 | 4 |
| PDHA1 | Pyruvate dehydrogenase E1 component α subunit, somatic form | Glycolysis | 30 | 3 |
| PMPCB | Mitochondrial processing peptidase β subunit | Mitochondrial organization | 31 | 4 |
| PRODH | Proline oxidase | Matrix | 25 | 6 |
| SOD2 | Superoxide dismutase [Mn] | Matrix | 23 | 2 |
| TIMM44 | Mitochondrial import inner membrane translocase subunit | Inner membrane | 20 | 4 |
Antibodies to mitochondrial proteins, among anti-keratinocyte antibodies and several other soluble pathogenic factors, act synergistically to activate keratinocyte cell death pathways in PV (Figure 1). In an organ culture of neonatal mouse skin—an in vitro model of PV—AMA and anti-Dsg1/3 autoantibodies acted synergistically to induce acantholysis [21]. Treatment with AMA alone did not result in acantholysis, however the combination of AMA and a mixture of anti-Dsg antibodies induced acantholysis. The AMA/anti-Dsg3 combination induced epidermal splitting suprabasally and the AMA/anti-Dsg1 combination induced epidermal splitting subcorneally, which is characteristic of pemphigus foliaceus. Moreover, while acantholysis was observed following treatment with high concentrations of the human anti-Dsg single-chain variable fragment (scFv), at low, physiologic doses the scFv was not able to induce keratinocyte detachment. Acantholysis was only observed when it was combined with AMA [21]. The synergy of AMA with other autoantibodies in PV implies that a simultaneous hit is required to alter the keratinocyte ability to maintain epidermal integrity. It is theorized that the binding of a single type of autoantibody only induces reversible changes in the keratinocyte, such that the cell maintains its ability to recover via self-repair mechanisms. Irreversible keratinocyte damage only occurs following presumed synchronized inactivation of salvage pathways by partnering autoantibodies, leading to loss of epidermal integrity [21].
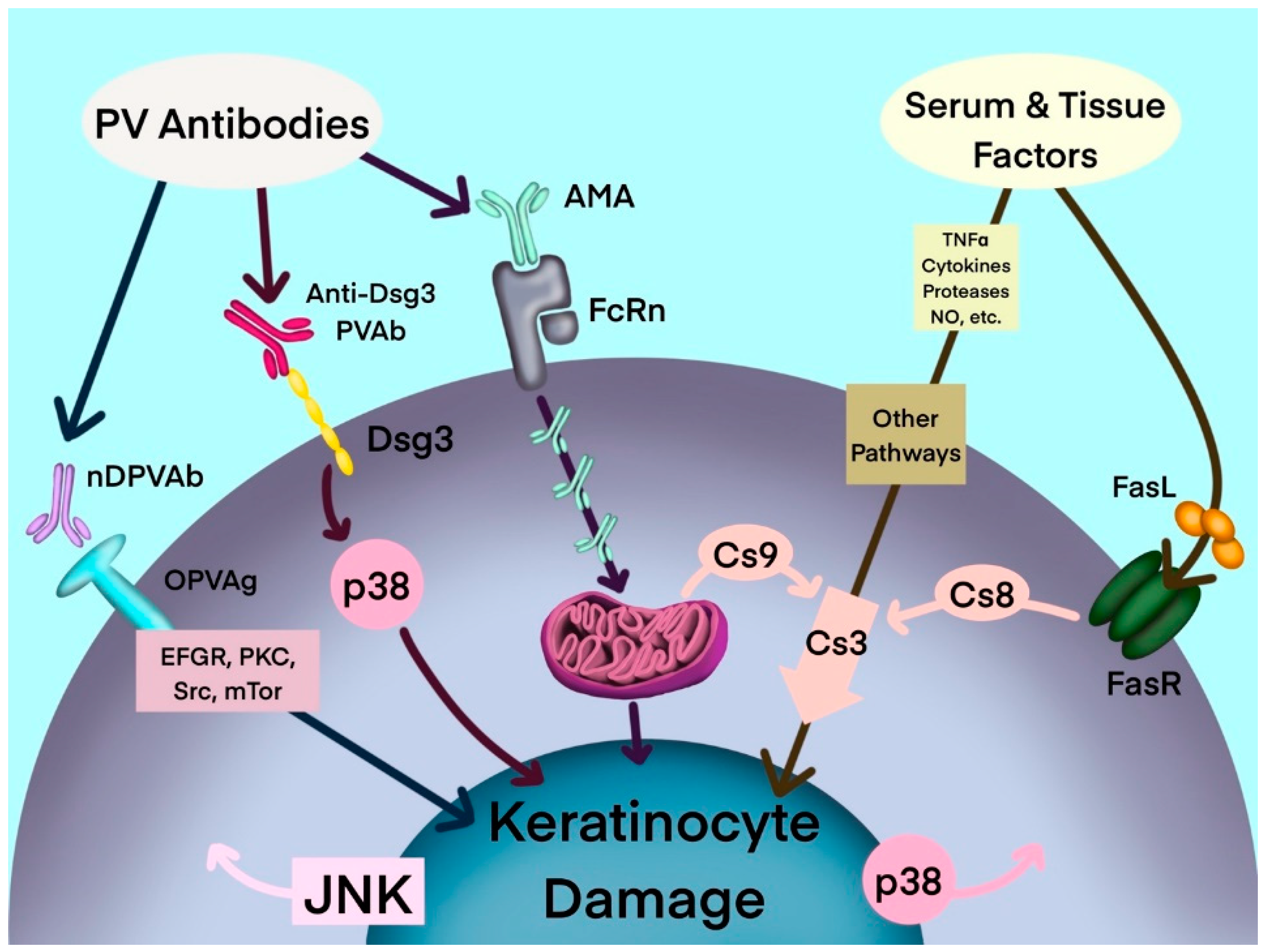
Figure 1. Hypothetical multipathogenic mechanism of interconnected signaling cascades leading to keratinocyte apoptolysis in pemphigus (modified from Marchenko et al. [19]). AMA: antimitochondrial antibodies; Anti-Dsg3 PVAb: anti-desmoglein 3 PV antibody; Cs: caspase; Dsg3: desmoglein-3; EGFR: epidermal growth factor receptor; FasL: Fas ligand; FasR: Fas receptor; JNK: c-Jun N-terminal kinase; mTOR: mammalian target of rapamycin; NO: nitric oxide; nDPVAb: non-Dsg PV antibodies; OPVAg: other PV antigens; PKC: protein kinase C; PVAb: PV antibody; Src: SRC proto-oncogene, nonreceptor tyrosine kinase; TNF-α: tumor necrosis factor-α.
This entry is adapted from the peer-reviewed paper 10.3390/antib11030055
References
- Lever, W.F.; Talbott, J.H. Pemphigus: A historical study. Arch. Dermatol. Syphilol. 1942, 46, 800–823.
- Dickson, S. Observations on Pemphigus. Lond. Med. J. 1788, 9, 309–324.
- Beutner, E.H.; Jordon, R.E. Demonstration of skin antibodies in sera of pemphigus vulgaris patients by indirect immunofluorescent staining. Proc. Soc. Exp. Biol. Med. 1964, 117, 505–510.
- Ahmed, A.R.; Carrozzo, M.; Caux, F.; Cirillo, N.; Dmochowski, M.; Alonso, A.E.; Gniadecki, R.; Hertl, M.; López-Zabalza, M.J.; Lotti, R.; et al. Monopathogenic vs. multipathogenic explanations of pemphigus pathophysiology. Exp. Dermatol. 2016, 25, 839–846.
- Grando, S.A. Pemphigus autoimmunity: Hypotheses and realities. Autoimmunity 2012, 45, 7–35.
- Amber, K.T.; Valdebran, M.; Grando, S.A. Non-Desmoglein Antibodies in Patients with Pemphigus Vulgaris. Front. Immunol. 2018, 9, 1190.
- Grando, S.A. Autoimmunity to keratinocyte acetylcholine receptors in pemphigus. Dermatology 2000, 201, 290–295.
- Chernyavsky, A.; Amber, K.T.; Agnoletti, A.F.; Wang, C.; Grando, S.A. Synergy among non-desmoglein antibodies contributes to the immunopathology of desmoglein antibody-negative pemphigus vulgaris. J. Biol. Chem. 2019, 294, 4520–4528.
- Sharma, P.; Mao, X.; Payne, A.S. Beyond steric hindrance: The role of adhesion signaling pathways in the pathogenesis of pemphigus. J. Dermatol. Sci. 2007, 48, 1–14.
- Grando, S.A.; Bystryn, J.C.; Chernyavsky, A.I.; Frušić-Zlotkin, M.; Gniadecki, R.; Lotti, R.; Milner, Y.; Pittelkow, M.R.; Pincelli, C. Apoptolysis: A novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp. Dermatol. 2009, 18, 764–770.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48.
- Gupta, S.; Kass, G.E.; Szegezdi, E.; Joseph, B. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033.
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516.
- Hasegawa, A.; Abe, R. Recent advances in managing and understanding Stevens-Johnson syndrome and toxic epidermal necrolysis . F1000Research 2020, 9, 612.
- Arredondo, J.; Chernyavsky, A.I.; Karaouni, A.; Grando, S.A. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in Pemphigus. Am. J. Pathol. 2005, 167, 1531–1544.
- Lotti, R.; Shu, E.; Petrachi, T.; Marconi, A.; Palazzo, E.; Quadri, M.; Lin, A.; O’Reilly, L.A.; Pincelli, C. Soluble Fas Ligand Is Essential for Blister Formation in Pemphigus. Front. Immunol. 2018, 9, 370.
- Kalantari-Dehaghi, M.; Chen, Y.; Deng, W.; Chernyavsky, A.; Marchenko, S.; Wang, P.H.; Grando, S.A. Mechanisms of mitochondrial damage in keratinocytes by pemphigus vulgaris antibodies. J. Biol. Chem. 2013, 288, 16916–16925.
- Marchenko, S.; Chernyavsky, A.I.; Arredondo, J.; Gindi, V.; Grando, S.A. Antimitochondrial autoantibodies in pemphigus vulgaris: A missing link in disease pathophysiology. J. Biol. Chem. 2010, 285, 3695–3704.
- Chernyavsky, A.; Chen, Y.; Wang, P.H.; Grando, S.A. Pemphigus vulgaris antibodies target the mitochondrial nicotinic acetylcholine receptors that protect keratinocytes from apoptolysis. Int. Immunopharmacol. 2015, 29, 76–80.
- Chen, Y.; Chernyavsky, A.; Webber, R.J.; Grando, S.A.; Wang, P.H. Critical Role of the Neonatal Fc Receptor (FcRn) in the Pathogenic Action of Antimitochondrial Autoantibodies Synergizing with Anti-desmoglein Autoantibodies in Pemphigus Vulgaris. J. Biol. Chem. 2015, 290, 23826–23837.
This entry is offline, you can click here to edit this entry!