Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The extracellular matrix (ECM) controls fundamental properties of tumors, including growth, blood vessel investment, and invasion. The ECM defines rigidity of tumor tissue and individual ECM proteins have distinct biological effects on tumor cells.
- TGFBI
- tenascin
- cancer-associated fibroblasts
- CAFs
- fibronectin
- collagen vi
- basement membrane
- interstitial collagen
- tumor
- matrix
1. Introduction
Several different primary tumors have been identified in the kidney; renal cell carcinoma (RCC) is the most common and one of the 10 most prevalent cancers overall [1]. In clear cell renal cell carcinoma (ccRCC), which is the most common subtype of RCC, clusters of tumor cells with clear cytoplasm are found in the kidney cortex [2]. The tumor suppressor VHL is lost in over 90% of ccRCCs through genetic or epigenetic mechanisms [3]. VHL plays a central role in cellular oxygen sensing [4][5], and its inactivation causes persistent pseudo-hypoxia, resulting in a strong angiogenic profile of tumors [6]. Evidence suggests that the cells of origin for ccRCC are epithelial cells that line the proximal tubule [7][8], one of the most metabolically active cell types in the human body [9]. Transformed epithelial cells in the tumor are surrounded by a network of stroma containing vasculature, interstitial fibroblasts and inflammatory and immune cells [10]. The cellular tumor components are embedded in the extracellular matrix (ECM).
Many fundamental properties of tumors are controlled by their ECM environment [11], including proliferation [12][13], vascularization [14][15] and invasion [16], making the ECM a key determinant of malignancy. Interplay between the ECM and tumor cells is complex and multifactorial. ECM provides the substrate for cancer cell attachment, and determines rigidity, which has a strong influence on malignancy [17]. It also controls signal transduction in cancer cells, exerting a strong influence on their behaviors [18].
The ECM is a mix of components with distinct physicochemical and signaling properties [19][20]. For the reasons outlined above, the ECM composition characteristic of a tumor type is predicted to play a central role in determining the behavior of that tumor. The ECM profile of a tumor is likely characteristic of the organ from which it arises, but also characteristic of the tumor’s cellular composition, with cancer-associated fibroblasts (CAFs) playing a major role in producing the tumor ECM [21]. Difficulties in isolating and maintaining CAFs have hampered experimental studies of these cells in ccRCC. The finding that the outgrowth of CAFs may be dependent on culture conditions that provide the complex ECM environment that they, themselves, generate highlights one important obstacle in the characterization of CAF biology [22]. Indeed, in vitro the ccRCC CAF population can be maintained by culturing it in an ECM environment that mimics the tumor of origin [23]. CAFs have features in common with activated myofibroblasts seen in fibrosis [24], and one interesting possibility is that insults that convert growth-suppressed fibroblasts in a healthy kidney to a proliferative and synthetic myofibroblast state may provide the appropriate environment for tumor formation from proximal tubule epithelial cells in which VHL has been mutated.
The Cancer Genome Atlas includes RNA-seq data for over 500 ccRCC tumors, along with information on patient outcomes, providing a popular resource for studies of association between gene expression and survival that may have prognostic value [25]. Using this resource, a gene signature has been identified that is comprised of 12 genes functionally associated with the ECM [26]. This dataset has also been used to derive a proxy signature for CAF investment in ccRCC, which anti-correlates with survival, suggesting that CAF infiltration in tumors is associated with poor prognosis [27]. While these correlative in silico studies cannot establish causality, they do suggest important relationships between ECM deposition, CAF abundance and tumor aggressiveness.
2. ECM Composition of ccRCC Tumors
Clinical material presents a complex matrix profile compared with cell line and short-term xenografting studies, since the tumor at the time of resection is perhaps decades old and composed of many different cell types. While the information that this type of material can yield regarding initiating events in the formation of the tumor is very limited, it does provide an important snapshot of the matrix environment at the time of surgery, and can potentially be correlated with invasiveness and metastasis. Additionally, recreating the matrix environment of the tumor is an important foundation for culturing primary tumor cells. The repertoire of ECM molecules in patient-derived ccRCC tumor samples was investigated with the goal of defining matrix components that could be defined as characteristic of ccRCC [23]. Seven stage pT3 tumors were collected from consenting patients undergoing partial nephrectomy along with healthy neighboring kidney cortex tissue. Neighboring tissue was designated “healthy” because it has a tissue layout and cell types characteristic of a healthy kidney cortex. Whether the cortex tissue neighboring the tumor is genuinely healthy or whether it is affected by proximity to the tumor is not known, and this is a general feature of studies that compare resected tumor tissue with healthy margins. The tumors and matched healthy cortices were analyzed via mass spectrometry using sequential window acquisition of all theoretical fragment ion spectra (SWATH) and data-dependent acquisition (DDA) modalities, which provided both a comparative analysis and a frequency table that could be used to infer protein abundance in tumor versus cortex. A defining feature of the ccRCC ECM composition characterized in this is its qualitative similarity to the healthy kidney cortex ECM. Within the detection limits of the analysis, neoplastic transformation does not lead to the de novo expression of matrix molecules, but rather, alters the relative abundance of components. One generalization that emerges from the analysis is that the abundance of basement membrane (BM) components is diminished in the ccRCC matrix, while the abundance of interstitial matrix (IM) components increases. Tissue from the healthy kidney cortex can be divided into functional units, which are nephrons and blood vessels, and the interstitial space between them. Nephrons and blood vessels are surrounded by BM, which insulates them from the IM scaffold in which renal fibroblasts are embedded (Figure 1). Considering the breakdown of normal epithelial structure that is characteristic of ccRCC, the abundance of IM and loss of BM seen in tumors is not surprising.
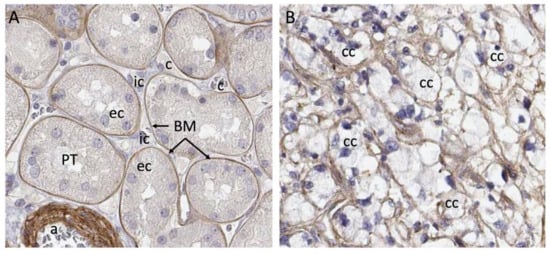
Figure 1. Breakdown of compartments in ccRCC. (A) Laminin a5 immunostaining shows strict basement membrane segregation of proximal tubule epithelial cells from the interstitium. (B) Cancer cells in ccRCC, which are thought to originate from the proximal tubule cell are embedded in matrix containing laminin a5, but there is no clear segregation of the interstitial cell space from the epithelium. Abbreviations: a—arteriole; BM—basement membrane; c—capillary; cc—clear cell; ec—epithelial cell; ic—interstitial cell. Images from Human Protein Atlas www.proteinatlas.org, licensed under the Creative Commons Attribution-ShareAlike 3.0 International License. https://www.proteinatlas.org/ENSG00000130702-LAMA5/tissue/kidney#img and https://www.proteinatlas.org/ENSG00000130702-LAMA5/pathology/renal+cancer#img were accessed on 12 August 2022.
In contrast to healthy cortex, ccRCC ECM is enriched in the IM components collagen VI, fibronectin, tenascin C, fibrin, TGFBI and periostin. Several studies indicate that these matrix components can influence the behavior of tumor cells, suggesting that the ECM environment could be an important determinant of tumor aggressiveness. Collagen VI is abundantly expressed in tumors from several organs, including breast [28], colon, and lung [29]. It promotes the survival of tumor cells [30] and fibroblasts [31] and has been shown to stimulate tumor angiogenesis [32]. The tumor content of collagen VI also promotes the invasive behavior of breast cancer cells [33] and colorectal cancer cells [34]. Studies of xenografted ccRCC cells show that collagen VI expression increases tumor size [35].
In summary, many lines of evidence support a role for collagen VI in promoting the aggressive behavior of tumor cells. Collagen VI is also an important driver of organ fibrosis and has been shown to directly promote the differentiation of cardiac fibroblasts to the hypersecretory myofibroblast phenotype, which is central to scarring after infarction [36]. The phenotypic conversion of vascular mural cells and fibroblasts to myofibroblasts is also central to fibrosis of the kidney [37], and the abundance of collagen VI in the ccRCC tumor may promote the development of a fibrotic local environment through the conversion of interstitial cells to myofibroblasts that secrete additional ECM components, the most characteristic being fibronectin [38]. Because collagen VI is a major component of the healthy kidney IM, it is difficult to argue that its presence in the tumor is sufficient to convert the matrix environment to a pro-fibrotic and pro-tumorigenic state. Rather, the contribution that collagen VI makes in the establishment of these pathologic environments may be contextual. Collagen VI is physically associated with a wide variety of other matrix proteins such as fibronectin [39], decorin [40][41], biglycan [41], fibulin [42], and collagens I [43] and IV [44], which were also identified as abundant components of ccRCC ECM [23]. The transition from healthy cortex to tumor tissue with its associated alterations in structure and the abundance of these proteins may generate a higher-order matrix complex combining BM and IM components that are normally sequestered in distinct tissue compartments. This matrix complex is predicted to have unique properties and may influence the behaviors of both tumor and stroma cells very differently from the healthy cortex BM and IM compartments.
Another possibility is that processing of the collagen VI peptide fragment endotrophin (ETP) increases in the tumor environment. ETP is a peptide generated by proteolytic cleavage of the carboxy-terminus of collagen VI α3 [45], normally occurring in the adipocytes of white adipose tissue. As fat mass increases, so does circulating ETP, and the finding that this “matrikine” acts as a driver of malignant tumor growth has led investigators to conclude that it may contribute to the association between obesity and cancer [46][47]. ETP is abundant in mammary tumors, which are surrounded by adipocytes [45]. Little is reported on the mechanism by which adipocytes release ETP from collagen VI α3, but one intriguing observation is that this mechanism is engaged when 3T3-L1 fibroblasts are chemically induced to accumulate lipid and form adipocytes [45]. It is possible that the ETP cleavage mechanism could also be activated in renal epithelial cells as they undergo the metabolic transformation to ccRCC tumor cells, in which fatty acid oxidation is repressed and lipids accumulate in the cytoplasm [48]. Alternately, ETP may be locally supplied by perirenal fat, the abundance of which has been correlated with cancer progression in ccRCC [49]. The increased relative risk of kidney cancer in obese individuals [50] strongly suggests a role for adiposity in ccRCC, and further investigation is required to understand whether ETP circulates to or is produced locally in ccRCC tumors.
Fibronectin can be found circulating as a soluble dimer, and in tissue as an insoluble ECM component [51]. Although fibronectin proteins are encoded by a single gene, functionally distinct isoforms are generated by alternate splicing. Circulating fibronectin produced by hepatocytes is largely devoid of the EIIIA and EIIIB domains present in the insoluble protein that is deposited in the tissue [52]. Fibronectin establishes fibrillar networks through integrin interactions, and these may be disturbed in VHL-inactivated ccRCC cells [53]. The understanding that fibronectin interacts with multiple other matrix molecules has led to a model of fibronectin as an organizer of higher-order matrix structure [54]. Together with its interacting partner collagen VI [39], it would be predicted to function as an adhesive for the majority of the most abundant matrix proteins identified in ccRCC tumors. Binding partners of particular relevance to the ccRCC matrisome include fibrin [55], periostin [56] and tenascin C [57]. Studies of cultured ccRCC cells in which fibronectin was knocked down suggest a role for fibronectin in promoting cell growth and migration in ccRCC [58]. The correlation of fibronectin expression with patient survival in the TCGA database shows an inverse relationship, suggesting detrimental effects of fibronectin in tumors [59]. A study of tumor tissue from 270 ccRCC patients that scored fibronectin protein expression in the membrane, cytoplasm and nucleus of tumor cells found higher disease-related mortality in patients with cytoplasmic fibronectin [60]. The genetic status of VHL was not defined in this patient cohort, and the impact of the loss of VHL-dependent fibronectin fibrillogenesis was not evaluated.
Tenascin C closely resembles fibronectin, shares receptor-binding properties and is expressed in the stroma of many solid tumors [61]. Little has been reported on the role of tenascin C in ccRCC, but a study of prognostic significance based on the correlation of clinical outcomes with histopathological evaluation of tumors from 137 patients showed that patients with tenascin C-positive tumors had a significantly lower survival rate and suggested that they also had increased risk of metastasis [62]. Studies in a model of glioblastoma have shown that tenascin C promotes proliferation and reduces cell adhesion by reducing the binding of fibronectin to its receptor syndecan 4, suggesting a cell biological mechanism for clinical correlation in ccRCC [63].
TGFBI is a TGFβ-induced protein that is secreted into the extracellular space [64] where it can bind to collagen VI [65] and fibronectin [66]. It shares almost 50% similarity with periostin and has been assigned roles as both a tumor suppressor and a tumor promoter in different experimental systems [67]. Studies in cultured ccRCC cells indicate that TGFBI promotes migration and invasion [68], suggesting a tumor-promoting role. A fluorescent in situ hybridization study of genes with predicted copy number variations in ccRCC confirmed copy number gain in TGFB1 and concluded that it may have a tumor-promoting role [69].
Periostin plays an important role in the organization of collagens by physically interacting with the enzymatic complex that covalently cross-links collagens and enhancing its activity [70]. It binds to both fibronectin and tenascin C [56], which are abundantly represented in the ccRCC ECM [23]. A study of tumor material from 1007 RCC patients concluded that elevated amounts of periostin in tumor cells were correlated with sarcomatoid differentiation and more aggressive tumor cell behavior [71].
Proteoglycans such as HSPG2 (perlecan), lumican and biglycan were identified in tumor ECM, although their abundance was similar to that in healthy cortex [23]. Proteoglycans are composed of a protein core to which glycosaminoglycan chains are covalently bound. HSPG2 is represented in the stroma of several tumor types [72], where it is predicted to bind tenascin C and modify growth factor signaling, including VEGF and FGF, by increasing the binding of these ligands to their receptors [73][74]. Lumican and biglycan are small leucine-rich proteoglycans (SLRPs) that promote collagen fibrillogenesis [75][76]. A study of 128 ccRCC patients, including 14 with matched primary ccRCC tumors and pulmonary metastases, correlated lumican expression with metastasis-free and overall patient survival [77].
Many of the studies reviewed suggest a connection between the abundance of specific ECM components, tumor aggressiveness and patient outcome. However, none of the ECM components reviewed are unique to ccRCC tumors, and all are represented in healthy kidney cortex where there is abundant representation of healthy tubule epithelial cells. One major difference between tumor tissue and the neighboring healthy cortex is that epithelial cells in the healthy cortex are insulated from the IM (Figure 1). In the tumor, the BM structure is broken down and these cells are exposed directly to the components of the IM. This may provide novel matrix signals that influence the process of transformation and tumor growth.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14174072
References
- Siegel, L.R.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Haake, M.S.; Rathmell, W.K. Renal cancer subtypes: Should we be lumping or splitting for therapeutic decision making? Cancer 2017, 123, 200–209.
- Nickerson, M.L.; Jaeger, E.; Shi, Y.; Durocher, J.A.; Mahurkar, S.; Zaridze, D.; Matveev, V.; Janout, V.; Kollarova, H.; Bencko, V.; et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin. Cancer Res. 2008, 14, 4726–4734.
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G., Jr.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275.
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530.
- Lindgren, D.; Sjölund, J.; Axelson, H. Tracing Renal Cell Carcinomas back to the Nephron. Trends Cancer 2018, 4, 472–484.
- Lindgren, D.; Eriksson, P.; Krawczyk, K.; Nilsson, H.; Hansson, J.; Veerla, S.; Sjolund, J.; Hoglund, M.; Johansson, M.E.; Axelson, H. Cell-Type-Specific Gene Programs of the Normal Human Nephron Define Kidney Cancer Subtypes. Cell Rep. 2017, 20, 1476–1489.
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646.
- Delahunt, B.; Srigley, J.R. The evolving classification of renal cell neoplasia. Semin. Diagn. Pathol. 2015, 32, 90–102.
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer 2021, 21, 217–238.
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997, 137, 231–245.
- Mettouchi, A.; Klein, S.; Guo, W.; Lopez-Lago, M.; Lemichez, E.; Westwick, J.K.; Giancotti, F.G. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol. Cell 2001, 8, 115–127.
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571.
- Brooks, P.C.; Montgomery, A.M.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164.
- Wang, Y.; McNiven, M.A. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J. Cell Biol. 2012, 196, 375–385.
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254.
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 2006, 126, 489–502.
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24.
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27.
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618.
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018, 67, 90–106.
- Bond, K.H.; Chiba, T.; Wynne, K.P.H.; Vary, C.P.H.; Sims-Lucas, S.; Coburn, J.M.; Oxburgh, L. The Extracellular Matrix Environment of Clear Cell Renal Cell Carcinoma Determines Cancer Associated Fibroblast Growth. Cancers 2021, 13, 5873.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49.
- Ahluwalia, P.; Ahluwalia, M.; Mondal, A.K.; Sahajpal, N.; Kota, V.; Rojiani, M.V.; Rojiani, A.M.; Kolhe, R. Prognostic and therapeutic implications of extracellular matrix associated gene signature in renal clear cell carcinoma. Sci. Rep. 2021, 11, 7561.
- Liu, B.; Chen, X.; Zhan, Y.; Wu, B.; Pan, S. Identification of a Gene Signature for Renal Cell Carcinoma-Associated Fibroblasts Mediating Cancer Progression and Affecting Prognosis. Front. Cell Dev. Biol. 2020, 8, 604627.
- Motrescu, E.R.; Blaise, S.; Etique, N.; Messaddeq, N.; Chenard, M.P.; Stoll, I.; Tomasetto, C.; Rio, M.C. Matrix metalloproteinase-11/stromelysin-3 exhibits collagenolytic function against collagen VI under normal and malignant conditions. Oncogene 2008, 27, 6347–6355.
- Nanda, A.; Carson-Walter, E.B.; Seaman, S.; Barber, T.D.; Stampfl, J.; Singh, S.; Vogelstein, B.; Kinzler, K.W.; St Croix, B. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI). Cancer Res. 2004, 64, 817–820.
- Iyengar, P.; Espina, V.; Williams, T.W.; Lin, Y.; Berry, D.; Jelicks, L.A.; Lee, H.; Temple, K.; Graves, R.; Pollard, J.; et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J. Clin. Investig. 2005, 115, 1163–1176.
- Rühl, M.; Sahin, E.; Johannsen, M.; Somasundaram, R.; Manski, D.; Riecken, E.O.; Schuppan, D. Soluble collagen VI drives serum-starved fibroblasts through S phase and prevents apoptosis via down-regulation of Bax. J. Biol. Chem. 1999, 274, 34361–34368.
- You, W.K.; Bonaldo, P.; Stallcup, W.B. Collagen VI ablation retards brain tumor progression due to deficits in assembly of the vascular basal lamina. Am. J. Pathol. 2012, 180, 1145–1158.
- Wishart, A.L.; Conner, S.J.; Guarin, J.R.; Fatherree, J.P.; Peng, Y.; McGinn, R.A.; Crews, R.; Naber, S.P.; Hunter, M.; Greenberg, A.S.; et al. Decellularized extracellular matrix scaffolds identify full-length collagen VI as a driver of breast cancer cell invasion in obesity and metastasis. Sci. Adv. 2020, 6, eabc3175.
- Liu, W.; Li, L.; Ye, H.; Tao, H.; He, H. Role of COL6A3 in colorectal cancer. Oncol. Rep. 2018, 39, 2527–2536.
- Wan, F.; Wang, H.; Shen, Y.; Zhang, H.; Shi, G.; Zhu, Y.; Dai, B.; Ye, D. Upregulation of COL6A1 is predictive of poor prognosis in clear cell renal cell carcinoma patients. Oncotarget 2015, 6, 27378–27387.
- Naugle, J.E.; Olson, E.R.; Zhang, X.; Mase, S.E.; Pilati, C.F.; Maron, M.B.; Folkesson, H.G.; Horne, W.I.; Doane, K.J.; Meszaros, J.G. Type VI collagen induces cardiac myofibroblast differentiation: Implications for postinfarction remodeling. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H323–H330.
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Patón, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286.
- Schiessl, I.M. The Role of Tubule-Interstitial Crosstalk in Renal Injury and Recovery. Semin. Nephrol. 2020, 40, 216–231.
- Tillet, E.; Wiedemann, H.; Golbik, R.; Pan, T.C.; Zhang, R.Z.; Mann, K.; Chu, M.L.; Timpl, R. Recombinant expression and structural and binding properties of alpha 1(VI) and alpha 2(VI) chains of human collagen type VI. Eur. J. Biochem. 1994, 221, 177–185.
- Bidanset, D.J.; Guidry, C.; Rosenberg, L.C.; Choi, H.U.; Timpl, R.; Hook, M. Binding of the proteoglycan decorin to collagen type VI. J. Biol. Chem. 1992, 267, 5250–5256.
- Wiberg, C.; Hedbom, E.; Khairullina, A.; Lamandé, S.R.; Oldberg, A.; Timpl, R.; Mörgelin, M.; Heinegård, D. Biglycan and decorin bind close to the n-terminal region of the collagen VI triple helix. J. Biol. Chem. 2001, 276, 18947–18952.
- Sasaki, T.; Göhring, W.; Pan, T.C.; Chu, M.L.; Timpl, R. Binding of mouse and human fibulin-2 to extracellular matrix ligands. J. Mol. Biol. 1995, 254, 892–899.
- Specks, U.; Nerlich, A.; Colby, T.V.; Wiest, I.; Timpl, R. Increased expression of type VI collagen in lung fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1956–1964.
- Kuo, H.J.; Maslen, C.L.; Keene, D.R.; Glanville, R.W. Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J. Biol. Chem. 1997, 272, 26522–26529.
- Park, J.; Scherer, P.E. Adipocyte-derived endotrophin promotes malignant tumor progression. J. Clin. Investig. 2012, 122, 4243–4256.
- Bu, D.; Crewe, C.; Kusminski, C.M.; Gordillo, R.; Ghaben, A.L.; Kim, M.; Park, J.; Deng, H.; Xiong, W.; Liu, X.Z.; et al. Human endotrophin as a driver of malignant tumor growth. JCI Insight 2019, 5, e125094.
- Park, J.; Scherer, P.E. Endotrophin—A novel factor linking obesity with aggressive tumor growth. Oncotarget 2012, 3, 1487–1488.
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769.
- Huang, H.; Chen, S.; Li, W.; Wu, X.; Xing, J. High perirenal fat thickness predicts a poor progression-free survival in patients with localized clear cell renal cell carcinoma. Urol. Oncol. 2018, 36, 157.e1–157.e6.
- Chow, W.H.; Dong, L.M.; Devesa, S.S. Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 2010, 7, 245–257.
- Yamada, K.M.; Kennedy, D.W. Fibroblast cellular and plasma fibronectins are similar but not identical. J. Cell Biol. 1979, 80, 492–498.
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011, 4, 21.
- Ohh, M.; Yauch, R.L.; Lonergan, K.M.; Whaley, J.M.; Stemmer-Rachamimov, A.O.; Louis, D.N.; Gavin, B.J.; Kley, N.; Kaelin, W.G., Jr.; Iliopoulos, O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol. Cell 1998, 1, 959–968.
- Zollinger, A.J.; Smith, M.L. Fibronectin, the extracellular glue. Matrix Biol. 2017, 60, 27–37.
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456.
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J. Biol. Chem. 2010, 285, 2028–2039.
- Tremble, P.; Chiquet-Ehrismann, R.; Werb, Z. The extracellular matrix ligands fibronectin and tenascin collaborate in regulating collagenase gene expression in fibroblasts. Mol. Biol. Cell 1994, 5, 439–453.
- Ou, Y.C.; Li, J.R.; Wang, J.D.; Chang, C.Y.; Wu, C.C.; Chen, W.Y.; Kuan, Y.H.; Liao, S.L.; Lu, H.C.; Chen, C.J. Fibronectin Promotes Cell Growth and Migration in Human Renal Cell Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2792.
- Majo, S.; Courtois, S.; Souleyreau, W.; Bikfalvi, A.; Auguste, P. Impact of Extracellular Matrix Components to Renal Cell Carcinoma Behavior. Front. Oncol. 2020, 10, 625.
- Steffens, S.; Schrader, A.J.; Vetter, G.; Eggers, H.; Blasig, H.; Becker, J.; Kuczyk, M.A.; Serth, J. Fibronectin 1 protein expression in clear cell renal cell carcinoma. Oncol. Lett. 2012, 3, 787–790.
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009, 3, 287–310.
- Ohno, Y.; Izumi, M.; Yoshioka, K.; Ohori, M.; Yonou, H.; Tachibana, M. Prognostic significance of tenascin-C expression in clear cell renal cell carcinoma. Oncol. Rep. 2008, 20, 511–516.
- Huang, W.; Chiquet-Ehrismann, R.; Moyano, J.V.; Garcia-Pardo, A.; Orend, G. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 2001, 61, 8586–8594.
- Skonier, J.; Neubauer, M.; Madisen, L.; Bennett, K.; Plowman, G.D.; Purchio, A.F. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol. 1992, 11, 511–522.
- Hanssen, E.; Reinboth, B.; Gibson, M.A. Covalent and non-covalent interactions of betaig-h3 with collagen VI. Beta ig-h3 is covalently attached to the amino-terminal region of collagen VI in tissue microfibrils. J. Biol. Chem. 2003, 278, 24334–24341.
- Billings, P.C.; Whitbeck, J.C.; Adams, C.S.; Abrams, W.R.; Cohen, A.J.; Engelsberg, B.N.; Howard, P.S.; Rosenbloom, J. The transforming growth factor-beta-inducible matrix protein (beta)ig-h3 interacts with fibronectin. J. Biol. Chem. 2002, 277, 28003–28009.
- Corona, A.; Blobe, G.C. The role of the extracellular matrix protein TGFBI in cancer. Cell Signal. 2021, 84, 110028.
- Shang, D.; Liu, Y.; Yang, P.; Chen, Y.; Tian, Y. TGFBI-promoted adhesion, migration and invasion of human renal cell carcinoma depends on inactivation of von Hippel-Lindau tumor suppressor. Urology 2012, 79, e1–e7.
- Matsuda, D.; Khoo, S.K.; Massie, A.; Iwamura, M.; Chen, J.; Petillo, D.; Wondergem, B.; Avallone, M.; Kloostra, S.J.; Tan, M.H.; et al. Identification of copy number alterations and its association with pathological features in clear cell and papillary RCC. Cancer Lett. 2008, 272, 260–267.
- Maruhashi, T.; Kii, I.; Saito, M.; Kudo, A. Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J. Biol. Chem. 2010, 285, 13294–13303.
- Morra, L.; Rechsteiner, M.; Casagrande, S.; Duc Luu, V.; Santimaria, R.; Diener, P.A.; Sulser, T.; Kristiansen, G.; Schraml, P.; Moch, H.; et al. Relevance of periostin splice variants in renal cell carcinoma. Am. J. Pathol. 2011, 179, 1513–1521.
- Cruz, L.A.; Tellman, T.V.; Farach-Carson, M.C. Flipping the Molecular Switch: Influence of Perlecan and Its Modifiers in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 133–146.
- Ishijima, M.; Suzuki, N.; Hozumi, K.; Matsunobu, T.; Kosaki, K.; Kaneko, H.; Hassell, J.R.; Arikawa-Hirasawa, E.; Yamada, Y. Perlecan modulates VEGF signaling and is essential for vascularization in endochondral bone formation. Matrix Biol. 2012, 31, 234–245.
- Zhou, Z.; Wang, J.; Cao, R.; Morita, H.; Soininen, R.; Chan, K.M.; Liu, B.; Cao, Y.; Tryggvason, K. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. 2004, 64, 4699–4702.
- Robinson, K.A.; Sun, M.; Barnum, C.E.; Weiss, S.N.; Huegel, J.; Shetye, S.S.; Lin, L.; Saez, D.; Adams, S.M.; Iozzo, R.V.; et al. Decorin and biglycan are necessary for maintaining collagen fibril structure, fiber realignment, and mechanical properties of mature tendons. Matrix Biol. 2017, 64, 81–93.
- Chen, S.; Young, M.F.; Chakravarti, S.; Birk, D.E. Interclass small leucine-rich repeat proteoglycan interactions regulate collagen fibrillogenesis and corneal stromal assembly. Matrix Biol. 2014, 35, 103–111.
- Ho, T.H.; Serie, D.J.; Parasramka, M.; Cheville, J.C.; Bot, B.M.; Tan, W.; Wang, L.; Joseph, R.W.; Hilton, T.; Leibovich, B.C.; et al. Differential gene expression profiling of matched primary renal cell carcinoma and metastases reveals upregulation of extracellular matrix genes. Ann. Oncol. 2017, 28, 604–610.
This entry is offline, you can click here to edit this entry!