Acute respiratory distress syndrome (ARDS) is a heterogeneous syndrome historically characterized by the presence of severe hypoxemia, high-permeability pulmonary edema manifesting as diffuse alveolar infiltrate on chest radiograph, and reduced compliance of the integrated respiratory system as a result of widespread compressive atelectasis and fluid-filled alveoli. Coronavirus disease 19 (COVID-19)-associated ARDS (C-ARDS) is a novel etiology caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that may present with distinct clinical features as a result of the viral pathobiology unique to SARS-CoV-2.
- COVID-19
- acute respiratory distress syndrome
- mechanical ventilation
1. Introduction
2. “Typical” ARDS
| Timing | Within 1 week of known clinical insult or new or worsening respiratory symptoms | |
| Chest imaging | Bilateral opacities on CXR or CT not fully explained by effusions, lobar/lung collapse, or nodules | |
| Origin of edema | Respiratory failure not fully explained by cardiac failure or fluid overload | |
| Oxygenation | Mild | 200 mm Hg < PaO2/FiO2 ≤ 300 mm Hg with PEEP or CPAP ≥ 5 cm H2O |
| Moderate | 100 mm Hg < PaO2/FiO2 ≤ 200 mm Hg with PEEP ≤ 5 cm H2O | |
| Severe | PaO2/FiO2 ≤ 100 mm Hg with PEEP ≥ 5 cm H2O | |
3. Viral Pathogenesis of SARS-CoV-2
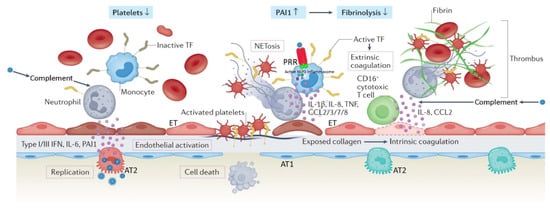
4. Distinct Pathologic Features of C-ARDS
Substantial clinical and biologic heterogeneity exists within the ARDS population [[60]]. Subphenotypes with distinct clinical features and responses to therapy have been identified with respect to the initial site of injury (pulmonary or extrapulmonary) [[61]] and biologic markers of inflammation (hypo- or hyperinflammatory) [[62]]. It should thus come as little surprise that properties unique to the SARS-CoV-2 virus itself might result in a form of ARDS with distinctive pathophysiology, or that even amongst patients with ARDS of a single etiology (e.g., C-ARDS), there might be a significant diversity of findings and responses to treatment (Table 2).
|
|
Classical ARDS |
C-ARDS |
|
Etiology |
Diverse, pulmonary or extrapulmonary (e.g. bacterial or viral pneumonia, severe trauma, aspiration, sepsis, etc.) |
SARS-COV-2 infection of alveolar type 2 cells (primarily) |
|
Hypoxemia (PaO2/FiO2 ≤300 mmHg at a PEEP level of ≥5 cmH2O) |
Acute onset (e.g. within <48 hours after the clinical insult), or progressive onset (i.e. within 7 days after the clinical insult) |
Progressive onset (i.e. within 7 or more days after the onset of COVID-19 symptoms)* |
|
Lung compliance at hypoxemia onset |
Usually low (e.g. <40 cmH2O/L) |
Usually high (e.g. >40 cmH2O/L) |
|
Recruitment potential |
Low or high, depending on the extent / nature of lung unit involvement and associated atelectasis |
Initially low – may increase with disease progression and development of edema and atelectasis |
|
Functional-to-anatomical shunt ratio / hyperperfusion of gasless tissue * |
Usually 0.5-2.0 / No |
Usually > 2.0 / Yes |
|
Alveolar capillary microthrombosis / new vessel growth |
Present / present |
Diffuse (~9 times more prevalent) / marked (2.7 times higher) |
|
Clinical benefit from lung-protective ventilation |
Proven |
Highly likely |
|
Clinical benefit from prone positioning |
Proven |
Highly likely |
|
Clinical benefit from corticosteroids |
Likely; more high-quality evidence needed |
Proven |
|
Clinical benefit from targeted anti-inflammatory interventions |
Uncertain; lack of intervention-specific evidence |
Proven |
|
Clinical benefit from ECMO |
Likely |
Possible; high-quality evidence still needed |
Table 2. Comparative presentation of major characteristic features of classical ARDS and C-ARDS. ARDS, acute respiratory distress syndrome; C-ARDS, coronavirus disease (COVID) 19-related ARDS; SARS-COV-2, severe acute, respiratory syndrome coronavirus 2; PaO2/FiO2, oxygen arterial partial pressure-to-fraction of inspired oxygen fraction ratio; PEEP, positive end-expiratory pressure; ECMO, extracorporeal membrane oxygenation. *, May predispose to early, profound hypoxemia and the conceptual risk of pre-intubation, patient self-inflicted lung injury.
Reports comparing the pathologic features of C-ARDS to other forms of viral- or non-viral ARDS are fraught with conflicting results, as accounting for the stage of disease and evolution of practice patterns over time is challenging. One theme that has consistently emerged, however, is the near-universal presence of pulmonary vascular abnormalities in patients with C-ARDS [[63]].
Though often present, pulmonary vascular lesions are not a dominant histopathologic feature of usual ARDS and are seldom widespread in post-mortem lung specimens [[64],[65]]. In patients with C-ARDS, however, they not only occur commonly [[66],[67]] but are extensive, occupying greater than 25% of the lung parenchyma in over half of the patients examined at autopsy in one study [[68]]. While microvascular thrombi may be a shared histologic finding among all patients with ARDS caused by pulmonary viruses, including Influenza A and SARS-CoV-1 [[69]], the extent of micro-thrombosis appears to be far greater in patients with C-ARDS [[70]]. This prevalence tends to uncouple gas exchange from mechanical properties, calling into question the specifics of ventilation management guidelines developed from clinical trials in the non-C-ARDS setting. Furthermore, the thrombotic burden is not confined to the microcirculation; the incidence of large-vessel pulmonary emboli is higher in patients with C-ARDS than in those of ARDS secondary to other viral and non-viral etiologies [[71],[72]]. Other pulmonary vascular derangements observed at autopsy include severe endothelial injury [[71],[72]] and the presence of dilated/engorged capillaries [[73]].
Studies incorporating dual-energy computerized tomographic angiography (CTA), digital subtraction CTA, and high-resolution CT have further extended these findings. Pulmonary vascular abnormalities on CT, most notably vessel enlargement, are common in patients with COVID-19 and may even be present prior to the development of C-ARDS [[74]]. Enlarged vessels suggestive of vasodilatation can be frequently observed within an area of ground glass or consolidation [[75]], contrary to the expected physiologic response to regional hypoxia (i.e., vasoconstriction). Perfusion imaging confirms that a considerable fraction of opacified lung parenchyma demonstrates increased uptake (indicating blood flow) in spite of diminished or even absent ventilation [[76]]. Perfusion abnormalities, on the other hand, are detected in areas of normal lung density [[74]], with one study of mechanically ventilated C-ARDS patients reporting that perfusion defects were not only present in every patient studied, but that the median extent of vascular abnormality approached 50% [[77]].
5. Respiratory Mechanics and Gas Exchange in C-ARDS
Early in the pandemic, Gattinoni and colleagues reported novel findings in their first 16 patients with C-ARDS; these patients had a relatively high tidal compliance (averaging 50.2 ml/cm H2O) associated with significantly elevated shunt fraction (0.50) [[78]]; furthermore, in the 8 patients they evaluated using quantitative CT, the ratio of shunt-fraction to gasless tissue was markedly higher (roughly 2.5 times) than those observed in usual ARDS [[79]], consistent with hyper perfusion of gasless tissue.
Chiumello and colleagues performed similar quantitative CT analysis in 32 consecutive C-ARDS patients receiving mechanical ventilation and compared gas exchange, respiratory mechanics, and CT variables to those of two historical cohorts of usual ARDS: one matched 1:1 for PaO2/FiO2 (P/F) and one matched 1:1 for compliance [[80]]. Compared to the C-ARDS cohort, the historical ARDS cohort matched for P/F had significantly lower compliance values (39.9 versus 49.4 mL/cmH2O) and gas volumes on CT (930 mL versus 1670 mL). The historical ARDS cohort matched for compliance, on the other hand, had a higher P/F when compared to the C-ARDS cohort (160 versus 106.5 mmHg).
These findings are well-explained by the pulmonary vasculopathy and diffuse, inflammation-triggered microthrombosis observed in COVID-related lung disease. In classical ARDS, airspace flooding, collapse, and consolidation tend to parallel the severity of oxygenation impairment and fall in compliance. C-ARDS challenges this conceptual framework; specifically, lung compliance may be well preserved in the early and mild stages of C-ARDS (at least in a major fraction of these patients), with severe hypoxemia not occurring primarily as a result of airspace filling and lung unit drop-out, but as the consequence of increased perfusion to non-ventilated lung units [[73],[81],[82],[83],[84]]. Over time, however, progression of C-ARDS fundamentally alters the lung’s mechanical properties. In late phase ARDS, regardless of the cause, lung capacity becomes severely reduced and is characterized by high dead space, limited recruitability, and low compliance [[85]].
As might be expected from the loosely defined and oxygenation-based criteria for ARDS and the evolving nature of COVID-related lung injury, there is wide overlap between the mechanics of C-ARDS and usual ARDS; indeed, several studies evaluating their comparative mechanical properties did not identify distinctive mean differences between cohorts [[86],[87]], which may in part be a function of the stage of illness in which such observations were made [[88],[89]].
6. Mechanical Ventilation in C-ARDS
The goals of invasive mechanical ventilation in C-ARDS are to relieve excessive work of breathing, improve gas exchange, and avoid aggravation of existing lung injury. Repeated exposure to tidal cycles that cause excessive, fracturing strain of structural microelements is believed to be the proximate mechanical stimulus for ventilator-induced lung injury (VILI) [[90]]; in recent years, a better understanding of the biophysical causes of VILI has shifted our traditional focus from the inflation pattern of a single tidal cycle toward avoiding exposure to damaging levels of tidal energy and power [[91]]. At the bedside, however, the focus remains on attempting to restrain tidal plateau and driving pressures below defined numerical thresholds. Unfortunately, this well-intentioned objective is often pursued through application of inflexible ventilatory targets and without consideration of the stage of disease.
In many patients with C-ARDS, ventilator strategies shown to be beneficial in clinical trials of unselected patients with ARDS will be appropriate; for others, however, they may not apply. The body of C-ARDS literature has expanded at a remarkable pace throughout the pandemic, providing guidance in certain areas regarding optimal ventilator management. Knowledge gained through physiologic studies preceding the C-ARDS era must be applied judiciously in order to provide individualized care for patients with ARDS of any etiology—including those with COVID-19.
7. Tidal Volume in C-ARDS
Twenty years ago the ARMA trial [9] demonstrated a 9% absolute reduction in mortality among mechanically ventilated ARDS patients randomized to an initial tidal volume of 6 mL/kg predicted body weight, forming the basis for what has become a standard of care codified in most ARDS guidelines [[92],[93]]. While large tidal volumes that lead to excessive strain are undoubtedly misguided in any acutely injured lung [[94]], several points are worth noting with respect to tidal volume selection in C-ARDS:
(1) Data from the ARMA trial, derived primarily from patients with ARDS secondary to bacterial pneumonia and sepsis, may not be wholly translatable to patients with ARDS secondary to novel forms of viral pneumonia with unique pathologic features, such as C-ARDS.
(2) Even in the ARMA trial, tidal volumes could be liberalized if necessary to facilitate patient comfort and adequate ventilation.
(3) In three large randomized trials that preceded the ARMA trial, no differences were found between patients treated with means of 7.2 mL/kg versus 10.6 mL/kg predicted body weight [[95]]; 7.2 mL/kg versus 10.4 ml/kg dry body weight [[96]]; and 7.3 ml/kg versus 10.2 ml/kg predicted body weight [[97]].
In the subpopulation of C-ARDS patients with less alveolar injury and relatively preserved compliance, larger tidal volumes of 7-8 mL/kg predicted body weight may result in tolerable strain and energy input without the risk of VILI [[91]]. In such patients, enforcing low tidal volumes can unnecessarily increase dead space [[98]], lead to reabsorption atelectasis from hypoventilation, and necessitate additional sedation to facilitate breathing comfort. However, as the severity of disease progresses and compliance declines, lower tidal volumes may be required to prevent the generation of strain that that exceeds critical thresholds of injury.
8. Application of PEEP in C-ARDS
Since the severity of gas exchange impairment and loss of compliance in the baby lung of ARDS reflect the reduced number of lung units available to accept ventilation, it is logical that interventions leading to an increase in the number of functional lung units should improve hypoxemia, reduce dead space, and increase compliance. Positive end-expiratory pressure (PEEP) is applied with the intent of achieving these goals by preventing collapse of unstable alveoli and thereby stabilizing “recruitment”. Expanding the ventilatory capacity in this manner additionally serves to distribute energy across a greater number of lung units, perhaps decreasing the quantity of damaging tidal energy transferred to the parenchymal matrix and reducing the risk of VILI [[19]].
Employing PEEP for the purposes of alveolar recruitment, however, hinges on the assumptions that compromised gas exchange is due primarily to loss of otherwise functional lung units and that these collapsed, or fluid-filled, units will regain function in response to the application of end-expiratory pressure. In C-ARDS, these assumptions may not hold true, and if they do, may be strongly dependent on the timing of the intervention [[99]].
Within the baby lung, the regional effects of PEEP are highly variable, as both recruitment and overdistension occur simultaneously as the lung expands. The net benefit of PEEP depends on whether recruitment of functional lung units outweighs overdistension within those that were already functional. When overdistension prevails, gas exchange is adversely affected as blood flow is directed away from overdistended lung units that previously participated in gas exchange, resulting in increased dead space and encouraging hypercarbia. The effects of net overdistension on oxygenation, on the other hand, are variable. Oxygenation may initially improve in response to increased PEEP despite net overdistension, especially if decreased cardiac output leads to reduction in blood flow through areas of intrapulmonary shunt, making the P/F ratio a poor surrogate for recruitment [[100],[101]].
When PEEP results in significant net recruitment, respiratory compliance (a correlate of baby lung size) will improve. However, when PEEP results in significant net overdistension, compliance will fall as open lung units are shifted past the upper inflection point of their pressure-volume curve. Under these conditions, the increased energy input associated with higher PEEP serves only to increase the risk of VILI and hemodynamic perturbations [[102]].
In recent decades, lung protective strategies have focused on not only the use of low tidal volumes for ventilation, but also the application of higher PEEP [[92]]. “PEEP tables,” in which PEEP is increased in a stepwise fashion with respect to the inspired oxygen requirement, assume that impaired oxygenation is secondary to the loss of functional lung units. Based on their use in clinical trials, such tables are commonly used by clinicians managing ARDS to select PEEP [[103]]. In many centers, this practice resulted in the early use of PEEP levels exceeding 14 cmH2O for C-ARDS [[104]]. In C-ARDS, however, impaired oxygen exchange is often strongly influenced by vascular dysfunction—not loss of functional lung units—in which case high levels of PEEP are not beneficial. In one study of mechanically ventilated patients with C-ARDS, partitioned respiratory mechanics were measured at low and high levels of PEEP [[105]]. Compared to 5 cmH2O, a PEEP of 15 cmH2O resulted in reduced lung compliance, increased lung strain, and an increased ventilatory ratio (i.e. a surrogate of physiological dead space defined as the quotient of measured over predicted product of minute ventilation and PaCO2 [[106]]). Had PEEP in that study [[105]] been set in accordance with the P/F table used in a recent clinical trial [[107]] it would have been 18 cmH2O.
While response to PEEP varies significantly among individual patients with C-ARDS [[100]], functional recruitment appears to be diminished relative to usual ARDS [[80]] and likely is influenced by the stage of disease and timing of observation [[108]]. Studies incorporating quantitative CT have either demonstrated minimal recruitment of additional lung units at higher levels of PEEP [[109]] or recruitment without simultaneous improvement in PaCO2, suggesting that recruited units are not functional/participating in gas exchange [[110]]. Indeed, higher levels of PEEP in C-ARDS have been reported to have deleterious effects on both gas exchange [[105],[111]] and respiratory mechanics [[105],[109],[111],[112],[113]], consistent with net overdistension. In the advanced stages of C-ARDS when consolidation is extensive, even PEEP levels as low as 5 cmH2O may be associated with markedly elevated airway plateau and driving pressures [[85]].
These data serve to underscore the importance of tailoring PEEP to the patient’s individual physiology. To minimize the hemodynamic and mechanical risks associated with PEEP, it should only be increased if doing so leads to demonstrable recruitment of functional lung units. While all methods of PEEP titration are imperfect, targeting optimal compliance is a reasonable strategy. If an increase in PEEP results in improved system compliance (while tidal volume is held constant), aeratable lung capacity has increased and recruitment has occurred. Recruitment of functional lung units is additionally associated with reduced PaCO2 for a given minute ventilation as a result of decreased dead space ventilation and increased surface area for gas exchange; while physiologic dead space is not routinely measured in clinical settings, the ventilatory ratio correlates reasonably well [[106]], is easily measured, and can be tracked following adjustments in PEEP. Similarly, the recruitment to inflation (R/I) ratio is a bedside test that has been used to estimate lung recruitability in response to changes in PEEP [[114]].
9. Body Positioning
Lung tissue mass is not distributed evenly, with 60% being located in the dependent (dorsal) half of the sterno-vertebral axis when supine [[115]]. In ARDS, the dorsal lung is predisposed to compressive atelectasis when supine due to the weight of overlying edematous tissue. External compression of lower lung units by the abdominal contents and of medial lung units by the weight of the overlying heart may also occur [[116],[117]]. Atelectasis results in relatively well-perfused but reversibly non-ventilated alveoli [[118]]. The ventral lung, on the other hand, is predisposed to overdistension during passive ventilation, not only because it receives a greater proportion of that ventilation, but also due to the increased regional compliance of the anterior chest wall (relative to the posterior chest wall), which permits a greater degree of end-tidal distension of adjacent lung units [[119]].
In the prone position, previously compressed dorsal and medial lung units are recruited, and previously gas-filled ventral lung units become less distended or collapse altogether. Despite this tendency for collapse of ventral lung units, there is typically net recruitment, as the loss of ventral lung units is outweighed by recruitment of units in the dorsal region, which contains a greater mass of lung tissue [[120]]. Prone positioning further results in better anatomical matching of the lung and chest wall shapes and compliance along the vertical axis, leading to less variation in size of individual pulmonary units [[119]] (Figure 2). Since the distribution of lung perfusion remains virtually unchanged in the prone position, these changes result in more homogenous ventilation, with decreases in both venous admixture and dead space. Proning may also result in reduced lung stress (i.e. transpulmonary pressure) and strain (i.e. the tidal volume-to-end-expiratory lung volume ratio) [[121]], decreasing the risk of VILI.
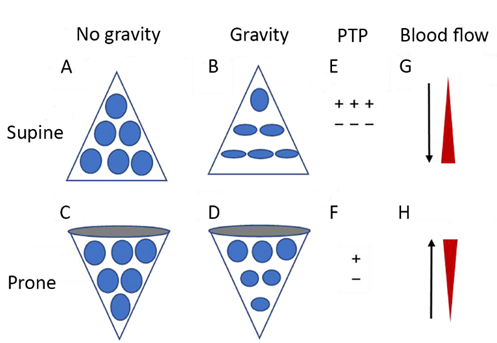
Figure 2. Diagrammatic presentation of physiological mechanisms associated with pronation in the acute respiratory distress syndrome (ARDS). (A) and (C) show the shape of lung units (i.e. alveoli) without the effect of gravity. (B) In the supine position, the volume of dorsal lung units is significantly smaller than the volume of ventral lung units, as a result of gravity and pleural pressure; thus, ventral lung units are more prone to overdistention and dorsal lung units are more prone to compression atelectasis. (D) In the prone position, gravity and pleural pressure result in a decrease in the volume of the ventral lung units and an increase in the volume of the dorsal lung units. (E) In the supine position, the ventral transpulmonary pressure (PTP) may substantially exceed the dorsal PTP. (F) Prone positioning reduces the ventral-to-dorsal PTP gradient, thereby augmenting the homogeneity of ventilation. (G) The reopening, dorsal lung units continue to receive most of the blood flow. (H) The ventral lung units may exhibit a greater tendency to collapse, but are still relatively underperfused. Reproduced in concordance with the Creative Commons Attribution License (CC-BY) from: Chen L, Zhang Y, Li Y, Song C, Lin F, Pan P. The Application of Awake-Prone Positioning Among Non-intubated Patients With COVID-19-Related ARDS: A Narrative Review. Front Med (Lausanne). 2022;9:817689.
The use of prone positioning has increased significantly during the COVID pandemic, with 77% of mechanically ventilated C-ARDS patients with a P/F < 100 being placed in the prone position [[122]] compared to only 16% of usual ARDS patients with a P/F < 100 during the pre-COVID era [[123]]. It remains one of the few interventions in severe ARDS associated with survival benefit, as demonstrated by a landmark study showing significant mortality reduction when patients with ARDS and a P/F < 150 were placed the prone position for least 16 hours daily [[124]]. While that trial preceded the advent of COVID, recent investigations performed in C-ARDS patients also suggest a survival benefit, with one retrospective study demonstrating a small but statistically significant reduction in the risk of death when C-ARDS patients with a P/F < 200 were proned within the first 2 days of ICU admission [[125]].
Studies that have investigated the physiologic effects of prone positioning in C-ARDS patients have generally reported improved oxygenation, with P/F increasing ≥ 20 mmHg in approximately 75% of patients [[122]]. Responses to proning are heterogeneous though, and available data suggest that the mechanisms responsible for improved oxygenation may differ from those in usual ARDS.
Unlike usual ARDS, net recruitment of C-ARDS lungs following placement in the prone position is relatively modest and often negligible [[126]]. Improved system compliance, typically present when significant net recruitment occurs, has not been observed in most studies [[122],[126],[127]-[128],[129]]. While measurements of partitioned respiratory mechanics would be needed to conclude with certainty that the lack of improvement in system compliance isn’t the result of decreased chest wall compliance in the prone position counterbalancing a simultaneous increase in compliance of newly recruited lung, an absence of significant recruitment is suggested by other findings as well.
CO2 exchange often improves in the prone position as a result of decreased dead space and recruitment of additional lung units. Most studies that have evaluated gas exchange in the prone position, however, have reported little change in the PaCO2 (or ventilatory ratio) [[122],[126],[127]-[128],[129]]. Compared to usual ARDS, the changes in both respiratory system compliance and PaCO2 following prone positioning are significantly less in patients with C-ARDS [[83]]. In the absence of recruitment, the most plausible mechanism to explain improved oxygenation is better matching of ventilation/perfusion ratios of vaso-dysregulated tissue [120]].
Timing may also play a significant role in response to prone positioning [[83],[126]]. In unresolving ARDS, atelectasis and edema may evolve into significant dorsal consolidation and diffuse fibrosis; in this setting, there is minimal recruitment of dorsal tissue in the prone position--only increased ventral atelectasis. A significant percentage of such patients either experience worsened P/F ratio in the prone position or fail to meet the accepted criteria for “responsiveness” (improvement in P/F ≥ 20 mmHg) [[128]].
10. Conclusion
Typical ARDS is characterized by high-permeability edema, widespread atelectasis, and a loss of compliance that relates directly to the reduced capacity of aerated lung units. COVID-19, a novel etiology of ARDS, has distinct pathologic findings consistent with severe injury to—and dysfunction of—the pulmonary vasculature as a result of SARS-CoV-2-induced endothelial injury and immunothrombosis. The lungs of patients with C-ARDS may be more likely to overdistend than to recruit in response to customary levels of PEEP. A subpopulation of patients with C-ARDS may present with severely deranged gas exchange that is uncoupled from the comparatively mild parenchymal injury. Just as typical ARDS encompasses a broad range of clinical findings, so too does C-ARDS, often transitioning in its more advanced stages to a form indistinguishable from typical ARDS. Some have argued that all patients with ARDS, regardless of etiology, should be treated identically. This approach, however, ignores the physiologic variability that not only exists between patients, but also within individual patients depending on the phase of the disease.
Randomized trials in ARDS have identified several interventions that lead to improved outcomes. These studies have enrolled patients with significant heterogeneity though and as such, a significant degree of heterogeneity in treatment effect is to be expected [[130]]. They report the mean intervention effects observed in a population, but with regard to benefit wide individual variability exists. Randomized trials have provided safe starting points from which to approach mechanical ventilation in the individual, but such rules are not inviolable. A more holistic approach, taking into consideration the unique physiology of individual patients, is warranted—as exemplified by C-ARDS.
This entry is adapted from the peer-reviewed paper 10.3390/jcm11164790
References
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323.
- Gattinoni, L.; Marini, J.J. Isn’t it time to abandon ARDS? The COVID-19 lesson. Crit. Care 2021, 25, 326.
- Bernard, G.R. Acute respiratory distress syndrome: A historical perspective. Am. J. Respir. Crit. Care Med. 2005, 172, 798–806.
- Thille, A.W.; Esteban, A.; Fernandez-Segoviano, P.; Rodriguez, J.M.; Aramburu, J.A.; Penuelas, O.; Cortes-Puch, I.; Cardinal-Fernandez, P.; Lorente, J.A.; Frutos-Vivar, F. Comparison of the Berlin definition for acute respiratory distress syndrome with autopsy. Am. J. Respir. Crit. Care Med. 2013, 187, 761–767.
- Marini, J.J. Limitations of clinical trials in acute lung injury and acute respiratory distress syndrome. Curr. Opin. Crit. Care 2006, 12, 25–31.
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533.
- Iuliano, A.D.; Brunkard, J.M.; Boehmer, T.K.; Peterson, E.; Adjei, S.; Binder, A.M.; Cobb, S.; Graff, P.; Hidalgo, P.; Panaggio, M.J.; et al. Trends in Disease Severity and Health Care Utilization During the Early Omicron Variant Period Compared with Previous SARS-CoV-2 High Transmission Periods—United States, December 2020–January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 146–152.
- Lim, Z.J.; Subramaniam, A.; Ponnapa Reddy, M.; Blecher, G.; Kadam, U.; Afroz, A.; Billah, B.; Ashwin, S.; Kubicki, M.; Bilotta, F.; et al. Case Fatality Rates for Patients with COVID-19 Requiring Invasive Mechanical Ventilation. A Meta-analysis. Am. J. Respir. Crit. Care Med. 2021, 203, 54–66.
- Acute Respiratory Distress Syndrome Network; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308.
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2017, 377, 1904–1905.
- Holm, B.A.; Matalon, S. Role of pulmonary surfactant in the development and treatment of adult respiratory distress syndrome. Anesth. Analg. 1989, 69, 805–818.
- Niklason, L.; Eckerstrom, J.; Jonson, B. The influence of venous admixture on alveolar dead space and carbon dioxide exchange in acute respiratory distress syndrome: Computer modelling. Crit. Care 2008, 12, R53.
- Radermacher, P.; Maggiore, S.M.; Mercat, A. Fifty Years of Research in ARDS. Gas Exchange in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2017, 196, 964–984.
- Robertson, H.T.; Swenson, E.R. What do dead-space measurements tell us about the lung with acute respiratory distress syndrome? Respir. Care 2004, 49, 1006–1007.
- Katzenstein, A.L.; Bloor, C.M.; Leibow, A.A. Diffuse alveolar damage—The role of oxygen, shock, and related factors. A review. Am. J. Pathol. 1976, 85, 209–228.
- Gattinoni, L.; Mascheroni, D.; Torresin, A.; Marcolin, R.; Fumagalli, R.; Vesconi, S.; Rossi, G.P.; Rossi, F.; Baglioni, S.; Bassi, F.; et al. Morphological response to positive end expiratory pressure in acute respiratory failure. Computerized tomography study. Intensive Care Med. 1986, 12, 137–142.
- Gattinoni, L.; Pelosi, P.; Vitale, G.; Pesenti, A.; D’Andrea, L.; Mascheroni, D. Body position changes redistribute lung computed-tomographic density in patients with acute respiratory failure. Anesthesiology 1991, 74, 15–23.
- Gattinoni, L.; Pesenti, A.; Avalli, L.; Rossi, F.; Bombino, M. Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am. Rev. Respir. Dis. 1987, 136, 730–736.
- Gattinoni, L.; Marini, J.J.; Pesenti, A.; Quintel, M.; Mancebo, J.; Brochard, L. The “baby lung” became an adult. Intensive Care Med. 2016, 42, 663–673.
- Gattinoni, L.; Pesenti, A. The concept of “baby lung”. Intensive Care Med. 2005, 31, 776–784.
- Gattinoni, L.; Pesenti, A.; Bombino, M.; Baglioni, S.; Rivolta, M.; Rossi, F.; Rossi, G.; Fumagalli, R.; Marcolin, R.; Mascheroni, D.; et al. Relationships between lung computed tomographic density, gas exchange, and PEEP in acute respiratory failure. Anesthesiology 1988, 69, 824–832.
- Marini, J.J.; Gattinoni, L. Time Course of Evolving Ventilator-Induced Lung Injury: The “Shrinking Baby Lung”. Crit. Care Med. 2020, 48, 1203–1209.
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793.
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278.
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284.
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.; van der Meer, Y.; Caly, L.; Druce, J.; de Vries, J.J.C.; Kikkert, M.; Barcena, M.; et al. SARS-coronavirus-2 replication in Vero E6 cells: Replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 2020, 101, 925–940.
- Khan, M.; Yoo, S.J.; Clijsters, M.; Backaert, W.; Vanstapel, A.; Speleman, K.; Lietaer, C.; Choi, S.; Hether, T.D.; Marcelis, L.; et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell 2021, 184, 5932–5949.e5915.
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e414.
- Osuchowski, M.F.; Winkler, M.S.; Skirecki, T.; Cajander, S.; Shankar-Hari, M.; Lachmann, G.; Monneret, G.; Venet, F.; Bauer, M.; Brunkhorst, F.M.; et al. The COVID-19 puzzle: Deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir. Med. 2021, 9, 622–642.
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036.
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316.
- Land, W.G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome-with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 2021, 22, 141–160.
- Hernandez Acosta, R.A.; Esquer Garrigos, Z.; Marcelin, J.R.; Vijayvargiya, P. COVID-19 Pathogenesis and Clinical Manifestations. Infect. Dis. Clin. N. Am. 2022, 36, 231–249.
- Sebag, S.C.; Bastarache, J.A.; Ware, L.B. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr. Pharm. Biotechnol. 2011, 12, 1481–1496.
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 2103–2109.
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418.
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128.
- Owens, A.P., 3rd; Mackman, N. Tissue factor and thrombosis: The clot starts here. Thromb. Haemost. 2010, 104, 432–439.
- Kenawy, H.I.; Boral, I.; Bevington, A. Complement-Coagulation Cross-Talk: A Potential Mediator of the Physiological Activation of Complement by Low pH. Front. Immunol. 2015, 6, 215.
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460.
- Bonaventura, A.; Vecchie, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329.
- Rodrigues, T.S.; de Sa, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707.
- Kambas, K.; Markiewski, M.M.; Pneumatikos, I.A.; Rafail, S.S.; Theodorou, V.; Konstantonis, D.; Kourtzelis, I.; Doumas, M.N.; Magotti, P.; Deangelis, R.A.; et al. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J. Immunol. 2008, 180, 7368–7375.
- Georg, P.; Astaburuaga-Garcia, R.; Bonaguro, L.; Brumhard, S.; Michalick, L.; Lippert, L.J.; Kostevc, T.; Gabel, C.; Schneider, M.; Streitz, M.; et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell 2022, 185, 493–512.e425.
- Ebeyer-Masotta, M.; Eichhorn, T.; Weiss, R.; Laukova, L.; Weber, V. Activated Platelets and Platelet-Derived Extracellular Vesicles Mediate COVID-19-Associated Immunothrombosis. Front. Cell Dev. Biol. 2022, 10, 914891.
- Mackman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation Abnormalities and Thrombosis in Patients Infected With SARS-CoV-2 and Other Pandemic Viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044.
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436.
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145.
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570.
- Severe Covid, G.G.; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534.
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98.
- Initiative, C.-H.G. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477.
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Maglorius Renkilaraj, M.R.L.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348.
- Koning, R.; Bastard, P.; Casanova, J.L.; Brouwer, M.C.; van de Beek, D.; Amsterdam, U.M.C. COVID-19 Biobank Investigators. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensive Care Med. 2021, 47, 704–706.
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585.
- Berlin, D.A.; Gulick, R.M.; Martinez, F.J. Severe Covid-19. N. Engl. J. Med. 2020, 383, 2451–2460.
- Kox, M.; Waalders, N.J.B.; Kooistra, E.J.; Gerretsen, J.; Pickkers, P. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA 2020, 234, 1565–1567.
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244.
- Wilson JG; COVID-19-Related ARDS: Key Mechanistic Features and Treatments. Crit Care 2020, 24, 102, 10.3390/jcm11164896.
- doi:10.1164/ajrccm.158.1.9708031; Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? . Am J Respir Crit Care Med 1998, 158, 3, doi:10.1164/ajrccm.158.1.9708031.
- Calfee CS; Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials.. Lancet Respir Med 2014, 2, 611, doi:10.1016/S2213-2600(14)70097-9.
- Satturwar S; Postmortem Findings Associated With SARS-CoV-2: Systematic Review and Meta-analysis.. Am J Surg Pathol 2021, 45, 587, doi:10.1097/PAS.0000000000001650.
- Katzenstein AL; Diffuse alveolar damage--the role of oxygen, shock, and related factors. A review. . Am J Pathol 1976, 85, 209, .
- Tomashefski JF; The pulmonary vascular lesions of the adult respiratory distress syndrome.. Am J Pathol 1983, 112, 112, .
- Hariri LP; Lung Histopathology in Coronavirus Disease 2019 as Compared With Severe Acute Respiratory Sydrome and H1N1 Influenza: A Systematic Review. . Chest 2021, 159, 73, 10.1016/j.chest.2020.09.259.
- Milross L; Post-mortem lung tissue: the fossil record of the pathophysiology and immunopathology of severe COVID-19.. Lancet Respir Med 2022, 10, 95, doi:10.1016/S2213-2600(21)00408-2.
- Carsana L.; Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. . Lancet Infect Dis 2020, 20, 1135, 10.1016/S1473-3099(20)30434-5.
- Satturwar S; Postmortem Findings Associated With SARS-CoV-2: Systematic Review and Meta-analysis.. Am J Surg Pathol 2021, 45, 87, doi:10.1097/PAS.0000000000001650.
- Ackermann M; Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med 2020, 383, 120, 10.1056/NEJMoa2015432.
- Poissy J; Pulmonary Embolism in Patients With COVID-19: Awareness of an Increased Prevalence. Circulation 2020, 142, 184, 10.1161/CIRCULATIONAHA.120.047430.
- Helms J; High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 2020, 46, 1089, 10.1007/s00134-020-06062-x.
- Villalba JA; Vasculopathy and Increased Vascular Congestion in Fatal COVID-19 and ARDS. Am J Respir Crit Care Med 2022, 10.1164/rccm.202109-2150OC, in press, 10.1164/rccm.202109-2150OC.
- Santamarina MG; COVID-19: What Iodine Maps From Perfusion CT can reveal-A Prospective Cohort Study. Crit Care 2020, 24, 19, doi:10.1186/s13054-020-03333-3.
- Li Q; CT features of coronavirus disease 2019 (COVID-19) with an emphasis on the vascular enlargement pattern. Eur J Radiol 2021, 134, 109442, 10.1016/j.ejrad.2020.109442.
- Poschenrieder F; Severe COVID-19 pneumonia: Perfusion analysis in correlation with pulmonary embolism and vessel enlargement using dual-energy CT data. PLoS One 2021, 16, e0252478, doi:10.1371/journal.pone.0252478.
- Patel BV; Pulmonary Angiopathy in Severe COVID-19: Physiologic, Imaging, and Hematologic Observations. Am J Respir Crit Care Med 2020, 202, 690, 10.1164/rccm.202004-1412OC.
- Gattinoni L; Anatomical and functional intrapulmonary shunt in acute respiratory distress syndrome. Crit Care Med 2008, 36, 669, 10.1097/01.CCM.0000300276.12074.E1.
- Gattinoni L; Anatomical and functional intrapulmonary shunt in acute respiratory distress syndrome. Crit Care Med 2008, 36, 669, 10.1097/01.CCM.0000300276.12074.E1.
- Chiumello D; Physiological and quantitative CT-scan characterization of COVID-19 and typical ARDS: a matched cohort study. Intensive Care Med 2020, 46, 2187, 10.1007/s00134-020-06281-2.
- Barbeta E; SARS-CoV-2-induced Acute Respiratory Distress Syndrome: Pulmonary Mechanics and Gas-Exchange Abnormalities. Ann Am Thorac Soc 2020, 17, 1164, 10.1513/AnnalsATS.202005-462RL.
- Vasques F; Physiological dead space ventilation, disease severity and outcome in ventilated patients with hypoxaemic respiratory failure due to coronavirus disease 2019. Intensive Care Med 2020, 46, 2092, 10.1007/s00134-020-06197-x..
- Camporota L; Prone Position in COVID-19 and -COVID-19 Acute Respiratory Distress Syndrome: An International Multicenter Observational Comparative Study. Crit Care Med 2022, 50, 633, 10.1097/CCM.0000000000005354.
- Grieco DL; Respiratory physiology of COVID-19-induced respiratory failure compared to ARDS of other etiologies. Crit Care 2020, 24, 529, 10.1186/s13054-020-03253-2.
- Kummer RL; Paradoxically Improved Respiratory Compliance With Abdominal Compression in COVID-19 ARDS. Chest 2021, 160, 1739, 10.1016/j.chest.2021.05.012.
- Haudebourg AF; Respiratory Mechanics of COVID-19- versus Non-COVID-19-associated Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2020, 202, 287, 10.1164/rccm.202004-1226LE.
- Panwar R; Compliance Phenotypes in Early Acute Respiratory Distress Syndrome before the COVID-19 Pandemic. Am J Respir Crit Care Med 2020, 202, 1244, 10.1164/rccm.202005-2046OC.
- Beloncle F; Longitudinal changes in compliance, oxygenation and ventilatory ratio in COVID-19 versus non-COVID-19 pulmonary acute respiratory distress syndrome. Crit Care 2021, 25, 248, 10.1186/s13054-021-03665-8.
- Gattinoni L.; Reply by Gattinoni et al. to Hedenstierna et al., to Maley et al., to Fowler et al., to Bhatia and Mohammed, to Bos, to Koumbourlis and Motoyama, and to Haouzi et al. Am J Respir Crit Care Med 2020, 202, 628, 10.1164/rccm.202004-1052LE.
- Marini JJ; Energetics and the Root Mechanical Cause for Ventilator-induced Lung Injury. Anesthesiology 2018, 128, 1062, doi:10.1097/ALN.0000000000002203.
- Marini JJ; Static and Dynamic Contributors to Ventilator-induced Lung Injury in Clinical Practice. Pressure, Energy, and Power. Am J Respir Crit Care Med 2020, 201, 767, 10.1164/rccm.201908-1545CI.
- Fan E; An Official American Thoracic Society/European Society of Intensive Care Medicine/Society of Critical Care Medicine Clinical Practice Guideline: Mechanical Ventilation in Adult Patients with Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2017, 195, 1253, 10.1164/rccm.201703-0548ST.
- Alhazzani W; Surviving Sepsis Campaign: Guidelines on the Management of Critically Ill Adults with Coronavirus Disease 2019 (COVID-19). Crit Care Med 2008, 28, e440, 10.1097/CCM.0000000000004363.
- Dreyfuss D; Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998, 157, 294, 10.1164/ajrccm.157.1.9604014.
- Stewart TE; Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. Pressure- and Volume-Limited Ventilation Strategy Group. N Engl J Med 1998, 338, 355, 10.1056/NEJM199802053380603.
- Brochard L; Tidal volume reduction for prevention of ventilator-induced lung injury in acute respiratory distress syndrome. The Multicenter Trail Group on Tidal Volume reduction in ARDS. Am J Respir Crit Care Med 1998, 158, 1831, 10.1164/ajrccm.158.6.9801044.
- Brower RG; Prospective, randomized, controlled clinical trial comparing traditional versus reduced tidal volume ventilation in acute respiratory distress syndrome patients. Crit Care Med 1999, 27, 1492, 10.1097/00003246-199908000-00015.
- Liu X; Ventilatory Ratio in Hypercapnic Mechanically Ventilated Patients with COVID-19-associated Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2020, 201, 1297, 10.1164/rccm.202002-0373LE.
- Marini JJ; Management of COVID-19 Respiratory Distress. JAMA 2020, 323, 2329, 10.1001/jama.2020.6825.
- Gattinoni L; Intensive Care Med . Intensive Care Med 2022, 48, 728, 10.1007/s00134-022-06698-x.
- Barthelemy R; Haemodynamic impact of positive end-expiratory pressure in SARS-CoV-2 acute respiratory distress syndrome: oxygenation versus oxygen delivery. Br J Anaesth 2021, 126, e70, 10.1016/j.bja.2020.10.026.
- Suter PM; Optimum end-expiratory airway pressure in patients with acute pulmonary failure. N Engl J Med 1975, 292, 284, 10.1056/NEJM197502062920604.
- Dickel S; Nationwide Cross-Sectional Online Survey on the Treatment of COVID-19-ARDS: High Variance in Standard of Care in German ICUs. J Clin Med 2021, 10, 3363, 10.3390/jcm10153363.
- Grasselli G; Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574, 10.1001/jama.2020.5394.
- Chiumello D; Positive end-expiratory pressure in COVID-19 acute respiratory distress syndrome: the heterogeneous effects. Crit Care 2021, 25, 431, 10.1186/s13054-021-03839-4.
- Sinha P; Analysis and Clinical Performance of the Ventilatory Ratio in Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2019, 199, 333, 10.1164/rccm.201804-0692OC.
- Beitler JR; Effect of Titrating Positive End-Expiratory Pressure (PEEP) With an Esophageal Pressure-Guided Strategy vs an Empirical High PEEP-Fio2 Strategy on Death and Days Free From Mechanical Ventilation Among Patients With Acute Respiratory Distress Syndrome: A Randomized Clinical Trial. JAMA 2019, 321, 846, 10.1001/jama.2019.0555.
- Pan C; Lung Recruitability in COVID-19-associated Acute Respiratory Distress Syndrome: A Single-Center Observational Study. Am J Respir Crit Care Med 2020, 201, 1294, 10.1164/rccm.202003-0527LE.
- Ball L; Computed tomography assessment of PEEP-induced alveolar recruitment in patients with severe COVID-19 pneumonia. Crit Care 2021, 25, 81, 10.1186/s13054-021-03477-w.
- Protti A; Lung Response to a Higher Positive End-Expiratory Pressure in Mechanically Ventilated Patients With COVID-19. Chest 2022, 161, 979, 10.1016/j.chest.2021.10.012.
- Roesthuis L; Advanced respiratory monitoring in COVID-19 patients: use less PEEP! . Crit Care 2020, 24, 230, 1186/s13054-020-02953-z.
- Perier F; Effect of Positive End-Expiratory Pressure and Proning on Ventilation and Perfusion in COVID-19 Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2020, 202, 1713, doi:10.1164/rccm.202008-3058LE.
- Mauri T; Potential for Lung Recruitment and Ventilation-Perfusion Mismatch in Patients With the Acute Respiratory Distress Syndrome From Coronavirus Disease 2019. Crit Care Med 2020, 48, 1129, 10.1097/CCM.0000000000004386.
- Chen L; Potential for Lung Recruitment Estimated by the Recruitment-to-Inflation Ratio in Acute Respiratory Distress Syndrome. A Clinical Trial. Am J Respir Crit Care Med 2020, 201, 178, 10.1164/rccm.201902-0334OC.
- Gattinoni L; Prone Position and COVID-19: Mechanisms and Effects. . Crit Care Med 2022, 50, 873, 10.1097/CCM.0000000000005486.
- Rouby J.J; Acute respiratory distress syndrome: lessons from computed tomography of the whole lung. Crit Care Med 2003, 31, 5285, 10.1097/01.CCM.0000057905.74813.BC.
- Albert RK; The prone position eliminates compression of the lungs by the heart. Am J Respir Crit Care Med 2000, 161, 1660, 10.1164/ajrccm.161.5.9901037.
- Pelosi P; Vertical gradient of regional lung inflation in adult respiratory distress syndrome. Am J Respir Crit Care Med 1994, 149, 8, 10.1164/ajrccm.149.1.8111603.
- Gattinoni L; Prone position in acute respiratory distress syndrome. Rationale, indications, and limits. Am J Respir Crit Care Med 2013, 188, 1286, 10.1164/rccm.201308-1532CI.
- Guerin C; Prone position in ARDS patients: why, when, how and for whom. Intensive Care Med 2020, 46, 2385, 10.1007/s00134-020-06306-w.
- Mentzelopoulos SD; Prone position reduces lung stress and strain in severe acute respiratory distress syndrome. Eur Respir J 2005, 25, 534, 10.1183/09031936.05.00105804.
- Langer T; Prone position in intubated, mechanically ventilated patients with COVID-19: a multi-centric study of more than 1000 patients. Crit Care 2021, 25, 128, 10.1186/s13054-021-03552-2.
- Bellani G; The LUNG SAFE study: a presentation of the prevalence of ARDS according to the Berlin Definition! . Crit Care 2016, 20, 268, 10.1186/s13054-016-1443-x.
- Guerin C; Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 2013, 368, 2159, 10.1056/NEJMoa1214103.
- Mathews KS; Prone Positioning and Survival in Mechanically Ventilated Patients With Coronavirus Disease 2019-Related Respiratory Failure. Crit Care Med 2021, 49, 1026, 10.1097/CCM.0000000000004938.
- Fossali T; Effects of Prone Position on Lung Recruitment and Ventilation-Perfusion Matching in Patients With COVID-19 Acute Respiratory Distress Syndrome: A Combined CT Scan/Electrical Impedance Tomography Study. Crit Care Med 2022, 50, 723, 10.1097/CCM.0000000000005450.
- Zarantonello F; Early physiological effects of prone positioning in COVID-19 Acute Respiratory Distress Syndrome. Anesthesiology 2022, 137, 327, 10.1097/ALN.0000000000004296.
- Rossi S; Mechanisms of oxygenation responses to proning and recruitment in COVID-19 pneumonia. Intensive Care Med 2022, 48, 56, 10.1007/s00134-021-06562-4.
- Protti A; Lung response to prone positioning in mechanically-ventilated patients with COVID-19.. Crit Care 2022, 26, 127, 10.1186/s13054-022-03996-0..
- Iwashyna TJ; Implications of Heterogeneity of Treatment Effect for Reporting and Analysis of Randomized Trials in Critical Care. Am J Respir Crit Care Med 2015, 192, 1045, 10.1164/rccm.201411-2125CP.