Genome-wide single nucleotide polymorphism (SNP) data are now quickly and inexpensively acquired, raising the prospect of creating personalized dietary recommendations based on an individual's genetic variability at multiple SNPs. In this review, we discuss the current potential for precision nutrition based on an individual's genetic data. We also review complicating issues and their impact on our ability to predict responses to dietary interventions.
- precision nutrition
- diet
- genetics
- SNP
- Modern Western Diet
- nutrigenomics
- genetic testing
- genomics
- metabolomics
- epigenetics
1. Introduction
Science in the 20th century yielded a basic understanding of the key macro and micro nutritional requirements for most humans. This resulted in a one-size-fits-all approach, exemplified by plans such as MyPlate and the Food Guide Pyramid, which have been impactful in reducing malnutrition and diseases resulting from nutrient deficiencies [1]. Precision nutrition, sometimes called personalized nutrition, nutrigenetics, or nutritional genetics, is the opposite—individuals receive diets tailored to their personal biology. Studies of global human genomic variation have demonstrated dramatic population-based differences in allele frequencies of common single nucleotide polymorphisms (SNPs) that influence the expression of genes responsible for the metabolism of some of the most common nutrients consumed by humans. In addition, evolutionary studies reveal that humans genetically adapted to their ancestral diets and local environments, as well as genetically drifted apart, giving rise to observed global patterns of sequence variation [2]. Consequently, individuals in large modern populations with diverse genetic ancestries such as the US may have a wide range of metabolic responses to the same food or diet, calling into question the one-size-fits-all dietary approach.
Diet-based genetic variation developed initially in Africa and continued as modern humans migrated out of Africa and across the globe over the past 100,000 years. Natural selection in response to new climates and food sources resulted in population- or region-specific genetic variation [3]. For example, the ability to digest lactose as an adult is much more common among Northern Europeans than East Asians or Africans [4,5]. In addition to these evolutionary studies, genome-wide association studies (GWAS) have discovered many genetic variants associated with specific nutrition-related traits including nutrient absorption, lipid metabolism, nutrient utilization, and fat accumulation that in turn can result in gene–diet interactions and human diseases. Together, these findings raise the critical question of whether dietary recommendations could be tailored to individuals based on genetic variation and how significant the impact of precision nutrition could be in contrast to conventional recommendations. Given the early nature of this science, it is not possible to adequately evaluate the overall effect of precision versus conventional nutrition. It is therefore our objective in this review to highlight both important examples in which genetic information can be helpful or vital in making nutritional recommendations and other examples in which it has limited value.
A growing number of companies now offer direct-to-consumer, genetically-based nutritional testing (DTC-GT) and advice [6]. The rapid growth of this industry is a testament to the fact that large numbers of consumers yearn for the purported benefits of “gene-based diets”. However, precision nutrition is at a very early stage and in most cases lacks sufficient science to be implemented, especially given the complexity of genetic alterations, and their effects, as well as a lack of knowledge of the dietary exposure necessary to induce a detrimental gene–diet interaction.
2. Genomic Architecture
The human genome consists of over 3 billion DNA base-pairs organized into chromosomes and present in the nuclei of most of our cells in two copies: one from each parent. It encodes the proteins our bodies need in linear units of information called genes, of which there are about 21,000 [7]. Genes occupy only a small fraction (<1%) of the genome; the rest includes “regulatory machinery”—regions that are important for controlling the transcription of various genes—as well as repetitive regions and large regions with unknown function(s) [7]. Transcriptional machinery “reads” the DNA code and produces mRNA. This mRNA then moves to ribosomes, where it interacts with translational machinery to link amino acids into the encoded proteins. Figure 1 shows an example of the organization of a typical gene, which consists of multiple regions referred to as exons and introns. While both are initially transcribed, spliceosomes remove the intronic regions, such that only exons are present in the mature mRNA transcripts. In addition, a single gene can produce multiple transcripts that each only contain certain exons, allowing single genes to encode multiple protein isoforms [8]. For reference, we provide a glossary of common genetic terms with simple explanations in Table 1.
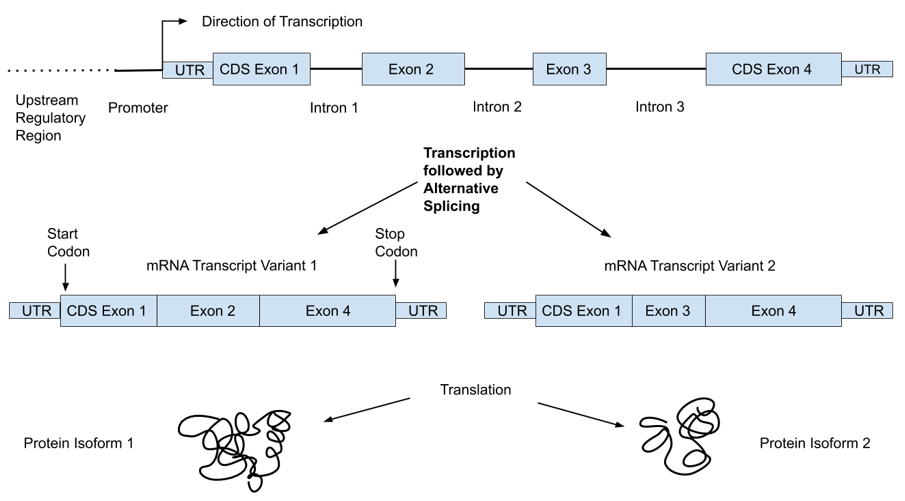
Figure 1. Basic architecture of a gene showing exons (eventually become the mature mRNA transcript), introns (removed during transcription), coding sequence regions within exons (CDS), and untranslated portions of exons (UTR).
Table 1. Glossary of common genetic terms.
|
Effect Size |
A measure of the size of a genetic association. Small effect sizes are common |
|
Epistasis |
When the effect of a variant depends on other genetic variants present (i.e., the genetic background) |
|
Genotype |
The two DNA bases at a given site, e.g., A/A, A/T or T/T, one from each parent |
|
Genotype-Phenotype Map |
The relationship between phenotypes and genotypes |
|
Heritability |
The degree to which a trait is transmitted across generations |
|
INDEL |
Insertion/deletion polymorphism |
|
Linkage Disequilibrium |
When nearby variants are passed down together through human lineages |
|
Locus |
A location in the genome |
|
Penetrance |
The probability of observing the associated phenotype for a given variant. |
|
Phenotype |
An observed trait, e.g., weight |
|
Pleiotropy |
When a single gene or variant controls multiple, sometimes unrelated traits |
|
Polygenic |
A phenotypic trait that is the result of small contributions from many genes |
|
Site |
A single DNA base-pair, i.e., A, C, G, or T, where the other half of the base-pair is implied |
|
Single nucleotide polymorphism (SNP) |
A site at which there are two common DNA base pairs in the population, e.g., A and T occur at 20 and 80% respectively |
|
Variant |
A DNA polymorphism, often a SNP |
|
Causal (functional) SNP |
A SNP that is responsible for the observed phenotypic association, e.g., a protein-altering mutation |
|
Dietary exposure |
The amount of a food or nutrient an individual or population consumes |
2.1. Genetic Variation
Despite being phenotypically quite diverse, humans are genetically mostly the same, with two individuals differing at <1% of their genomes on average (https://www.ncbi.nlm.nih.gov/books/NBK20363/). There are multiple ways two genomes can differ, and the simplest and most widely studied type of genetic variation is single base pair differences known as single nucleotide polymorphisms (SNPs). Other types of variation include insertions and deletions of short DNA fragments (INDELs); copy number variants (CNV), where a given gene is present in multiple copies and that number varies by individual; and structural variants (SVs), where larger genomic rearrangements exist. Although our knowledge of INDELs, CNVs, and SVs is growing, most of the nutritional genomics research to date has focused on SNPs, and those are the central focus for this review as well. The Genome Reference Consortium (https://www.ncbi.nlm.nih.gov/grc) helps to maintain a regularly updated human reference genome, and variation herein is described in relation to that reference.
Since the human genome was first sequenced in 2003, several large international projects such as HapMap [9], the Human Genome Diversity Panel [10], and the 1000 Genomes Project [11] have worked to sequence the genomes of thousands of individuals from around the world, and have created large catalogues of human genetic variation. We now know that SNPs exist about every 1000 bp, and over 300 million SNPs have been found to date [12]. The popular dbSNP database (https://www.ncbi.nlm.nih.gov/snp/), hosted by the National Center for Biotechnology Information, currently contains information on over 100 million SNPs (National Center for Biotechnology Information, U.S. National Library of Medicine).
2.2. From Genotype to Phenotype
Although the central dogma of molecular biology is that the progression from DNA to RNA to protein is straightforward, multiple changes can occur along that process that alter the expression of genes, and in turn the influence the effect of any genetic variant. While a detailed discussion of gene expression or epigenetics is beyond the scope of this review, an important point is that individual variants may not be expressed equally in all individuals. Determining SNP genotypes is straightforward but understanding the complex molecular and metabolic network of events impacted by an individual variant is far more difficult. Some SNP sites have known functions or associations with diseases or other phenotypic characteristics, including metabolism of dietary components and nutritional deficiencies, but these variants are the exception and not the rule. Moreover, in cases where a clinical association has been established, these relationships may not apply to different racial/ethnic populations. Further, many traits have strong developmental and environmental components and relatively low heritability. The lower part of Figure 1 shows how a single gene can result in multiple proteins, which are often expressed in different tissues or developmental stages.
In fact, most associated SNPs are not the functional SNPs, but rather in close linkage with other variants (perhaps containing the causal [functional] SNP) in the same region of the chromosome [13]. In this case, the associated SNP and the casual SNP are simply passed down together through human lineages (a phenomenon known as linkage disequilibrium) until they are separated by a relatively rare recombination event [13]. It is well recognized that different ancestry groups have varying degrees of linkage disequilibrium, and, thus, an association found in one population may not be valid for a population where it has not yet been established because the linkage between the marker SNP and the true causal variant may have been disrupted by a recombination event on the branch leading to that population [13].
2.3. Penetrance, Pleiotropy, Epistasis, and Polygenicity
Most traits of interest are complex, and several other genetic concepts help to explain the genotype-to-phenotype map. Penetrance is the probability of observing a trait, given that an individual has the associated variant or genotype [14]. A fully penetrant variant would be one such that everyone who had it also had the associated phenotype. It is important because in many cases having a particular genetic variant does not definitively result in the associated phenotype. Instead, it increases (or decreases) the chance of expressing that phenotype. Further, many traits are likely highly polygenic, that is, the observed phenotype is the results of contributions from many individual genes. For example, ~60 and ~100 independent loci contribute to the genetic risk associated with coronary artery disease and type 2 diabetes, respectively, with each individual locus contributing only a small effect on the underlying disease [15,16]. Some have attempted genetic risk scores (GRS) that examine the effects of multiple SNPs simultaneously; however, these scores often account for only a small proportion of the total trait variance. For example, when Vallée Marcotte et al. examined the triglyceride response to omega-3 supplementation, they found that a GRS with the top five SNPs accounted for only 11% of the total trait variance [17].
Single genes can also have multiple effects. Pleiotropy occurs when one gene is related to several different and often unrelated traits [18]. For example, sickle cell anemia is a disease caused by pleiotropy where a single gene mutation results in intense interindividual differences in the severity of the disease [19]. Epistasis occurs when the effect of one variant is dependent on the presence of other genetic variants; therefore, the full genetic architecture of the individual is important [20]. For example, SNPs in ACE, FTO, MC4R, and PPARG have all individually been associated with BMI, but, in an Italian cohort, Bordoni et al. found that the ACE variant appears protective against negative consequences of the MC4R variant [21]. In sickle cell anemia, several epistatic, pleiotropic genes (including genes that express adherence proteins, red cell receptors, and white cells) have been defined in the last decade, and many others are potential candidates [19].
In the context of nutrigenetics, polygenicity, pleiotropy, and epistasis all complicate the translation of genetic research into dietary recommendations. Phenotypic traits such as obesity, cholesterol, or plasma triglycerides have large numbers of associated variants. Consequently, it is difficult to predict what the impact of combinations, or how one variant may alter complex gene–diet interactions or other associations.
2.4. Genome-Wide Genetic Association Studies (GWAS)
Discerning the biological effects of the enormous catalogue of human variation is challenging. GWAS are based on the common-disease common-variant (CD-CV) hypothesis that common disease-causing alleles will underlie many common human diseases. They have played important roles in our understanding of many diseases and have identified many loci that are associated with various phenotypic traits. The basic design of GWAS studies is to take cases and controls for a given trait and to genotype several hundred thousand to a few million SNPs across the genomes of all subjects. For each SNP, a regression is performed, and a p-value obtained. After adjusting for multiple testing, one can then plot the p-values (often −log[p-values]) across the genome in a Manhattan plot to identify the sites that are most associated with a given trait, which appear as peaks (“skyscrapers”) in the graph. The sample sizes for a GWAS typically need to be very large—often in the 1000s—due to the very large number of SNPs that are tested.
This approach has been very popular and the GWAS catalog has grown to include over 188,000 SNP–trait associations (https://www.ebi.ac.uk/gwas/). Examples relevant to personalized nutrition include SNPs related to fasting blood glucose, macronutrient and micronutrient intake metabolism, cholesterol metabolism, obesity, vitamin D levels, and many other diet-related traits. Unfortunately, most GWAS have predominantly been performed in individuals of European ancestry, and the results found in one population do not always generalize to other populations [22]. Another key aspect of these studies is that they are typically hypothesis-generating, i.e., it is generally unknown which genes will emerge. For this reason, GWAS results should always be viewed as preliminary, in need of follow-up in additional and diverse cohorts. Even when a result has been validated in multiple cohorts, an understanding of its significance typically requires functional studies focused on how gene and protein expression as well as metabolic networks are affected.
References
- Kim, J.W. Direct-to-consumer genetic testing. Inf. 2019, 17, e34, doi:10.5808/GI.2019.17.3.e34.
- Luca, F.; Perry, G.H.; Di Rienzo, A. Evolutionary adaptations to dietary changes. Rev. Nutr. 2010, 30, 291–314, doi:10.1146/annurev-nutr-080508-141048.
- Fan, S.; Hansen, M.E.; Lo, Y.; Tishkoff, S.A. Going global by adapting local: A review of recent human adaptation. Science 2016, 354, 54–59, doi:10.1126/science.aaf5098.
- Heine, R.G.; Alrefaee, F.; Bachina, P.; de Leon, J.C.; Geng, L.; Gong, S.; Madrazo, J.A.; Ngamphaiboon, J.; Ong, C.; Rogacion, J.M. Lactose intolerance and gastrointestinal cow’s milk allergy in infants and children—common misconceptions revisited. World Allergy J. 2017, 10, 41, doi:10.1186/s40413-017-0173-0.
- Lapides, R.A.; Savaiano, D.A. Gender, age, race and lactose intolerance: Is there evidence to support a differential symptom response? A scoping review. Nutrients 2018, 10, doi:10.3390/nu10121956.
- Caulfield, T.; Ries, N.M.; Ray, P.N.; Shuman, C.; Wilson, B. Direct-to-consumer genetic testing: Good, bad or benign? Genet. 2010, 77, 101–105, doi:10.1111/j.1399-0004.2009.01291.x.
- Salzberg, S.L. Open questions: How many genes do we have? BMC 2018, 16, 94, doi:10.1186/s12915-018-0564-x.
- Larochelle, S. Protein isoforms: More than meets the eye. Methods 2016, 13, 291, doi.org/10.1038/nmeth.3828.
- Gibbs, R.A.; Belmont, J.W.; Hardenbol, P.; Willis, T.D.; Yu, F.; Yang, H.; Ch’ang, L.Y.; Huang, W.; Liu, B.; Shen, Y.; et al. The International HapMap Project. Nature 2003, 426, 789–796, doi:10.1038/nature02168.
- Li, J.Z.; Absher, D.M.; Tang, H.; Southwick, A.M.; Casto, A.M.; Ramachandran, S.; Cann, H.M.; Barsh, G.S.; Feldman, M.; Cavalli-Sforza, L.L.; et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008, 319, 1100–1104, doi:10.1126/science.1153717.
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74, doi:10.1038/nature15393.
- Nelson, M.R.; Marnellos, G.; Kammerer, S.; Hoyal, C.R.; Shi, M.M.; Cantor, C.R.; Braun, A. Large-scale validation of single nucleotide polymorphisms in gene regions. Genome 2004, 14, 1664–1668, doi:10.1101/gr.2421604.
- Clark, A.G.; Nielsen, R.; Signorovitch, J.; Matise, T.C.; Glanowski, S.; Heil, J.; Winn-Deen, E.S.; Holden, A.L.; Lai, E. Linkage disequilibrium and inference of ancestral recombination in 538 single-nucleotide polymorphism clusters across the human genome. J. Hum. Genet. 2003, 73, 285–300, doi.org/10.1086/377138.
- Wright, C.F.; West, B.; Tuke, M.; Jones, S.E.; Patel, K.; Laver, T.W.; Beaumont, R.N.; Tyrell, J.; Wood, A.R.; Frayling, T.M.; et al. Assessing the pathogenicity, penetrance, and expressivity of putative disease-causing variants in a population setting. J. Hum. Genet. 2019, 104, 2, 275–286, doi:10.1016/j.ajhg.2018.12.015.
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Genet. 2015, 47, 1121–1130, doi:10.1038/ng.3396.
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47, doi:10.1038/nature18642.
- Marcotte, B.V.; Guénard, F.; Marquis, J.; Charpagne, A.; Vadillo-Ortega, F.; Tejero, M.E.; Binia, A.; Vohl, M.-C. Genetic Risk Score Predictive of the Plasma Triglyceride Response to an Omega-3 Fatty Acid Supplementation in a Mexican Population. Nutrients 2019, 11, 737, doi:10.3390/nu11040737.
- Solovieff, N.; Cotsapas, C.; Lee, P.H.; Purcell, S.M.; Smoller, J.W. Pleiotropy in complex traits: Challenges and strategies. Rev. Genet. 2013, 14, 483–495, doi:10.1038/nrg3461.
- Nagel, R.L. Pleiotropic and epistatic effects in sickle cell anemia. Opin. Hematol. 2001, 8, 105–110, doi:10.1097/00062752-200103000-00008.
- Moore, J.H.; Williams, S.M. Epistasis and its implications for personal genetics. J. Hum. Genet. 2009, 85, 309–320, doi:10.1016/j.ajhg.2009.08.006.
- Bordoni, L.; Marchegiani, F.; Piangerelli, M.; Napolioni, V.; Gabbianelli, R. Obesity-related genetic polymorphisms and adiposity indices in a young Italian population. IUBMB Life 2017, 69, 98–105, doi:10.1002/iub.1596.
- Medina-Gomez, C.; Felix, J.F.; Estrada, K.; Peters, M.J.; Herrera, L.; Kruithof, C.J.; Duijts, L.; Hofman, A.; van Duijn, C.M.; Uitterlinden, A.G.; et al. Challenges in conducting genome-wide association studies in highly admixed multi-ethnic populations: The Generation R Study. J. Epidemiol. 2015, 30, 317–330, doi:10.1007/s10654-015-9998-4.
This entry is adapted from the peer-reviewed paper 10.3390/nu12103118