1. Introduction
Humans live in cooperation with their gut microbiome, as an integrated superorganism [
1,
2]. More than 100 trillion microorganisms, including over 1000 species of bacteria, archaea, viruses, fungi, and protozoa, are hosted in our gastrointestinal tract and are now recognized as the variable part of our genome [
3,
4]. The composition of the gut microbiome is the final result of the interplay between a complex network of factors, including the genetic landscape and environmental agents, immune response, and dietary habits [
5]. Beyond its critical role in many metabolic pathways [
6], the gut microbiome is involved in the maintenance of the intestinal barrier’s integrity, the protection of the host against pathogens, and the regulation of both innate and adaptive host immunity [
7]. A perturbation of this balance results in dysbiosis, a condition that can contribute to the pathogenesis and the further evolution of different disorders, including liver diseases [
8].
The gut–liver axis is an entity that stems from the close anatomical and functional relationship between the gastrointestinal tract and the liver [
9]. Under physiological conditions, this system allows only a small amount of bacteria and their products to reach the liver through portal circulation, where they are readily eliminated. In this way, the hepatic firewall prevents the dissemination of potential inflammatory triggers into the systemic bloodstream, maintaining a balanced immune response [
10]. With the development and progression of liver disease, the gut–liver axis undergoes a gradual and profound change, characterized by a breakdown of the intestinal barrier, dysbiosis, bacterial overgrowth, and excessive bacterial translocation. This causes a dysfunctional immune response perpetuating hepatic and systemic inflammation, which worsens liver damage into a vicious cycle [
11].
It is, therefore, not surprising that pathological changes in the gut microbiome have been associated with advanced chronic liver disease (ACLD) and its complications, such as hepatic encephalopathy, spontaneous bacterial peritonitis, and hepatocellular carcinoma [
12]. In recent years, increasing attention has been paid to the role that portal hypertension plays in shaping the gut–liver axis [
13,
14]. Since portal hypertension represents the primary driver of hepatic decompensation, which, in turn, is associated with increased mortality and morbidity in cirrhotic patients, a proper understanding of its link with the gut microbiome is of paramount importance for diagnostic, prognostic, and therapeutic approaches [
15]. Indeed, the last Baveno VII consensus in portal hypertension underlined the importance of the gut microbiome as one of the fields that needs to be explored in the future in order to improve the management of portal hypertension in patients with ACLD [
16].
2. Pathogenesis of Portal Hypertension in Liver Disease
Portal hypertension represents one of the major consequences of ACLD; it is defined as an increase in the hepatic venous pressure gradient (HVPG) of >5 mmHg [
16]. Clinically significant portal hypertension (CSPH) develops in the case of HVPG > 10 mmHg and is related to all of the complications of ACLD, such as gastroesophageal variceal bleeding, hepatic encephalopathy, and ascites. These complications represent the first cause of death and the main indication for liver transplantation in these patients [
17].
The development of portal hypertension in ACLD results from both an increased inflow and an obstructed outflow in the hepatic venous system. Indeed, structural modifications of hepatic sinusoids due to fibrosis and regenerative nodules, together with vasoconstriction in the intrahepatic circulation due to decreased production of vasodilators from sinusoidal endothelial cells, are responsible for the rise in intrahepatic vascular resistance [
18,
19]. On the other hand, splanchnic vasodilation, as a consequence of the huge amount of nitric oxide (NO) released by hyperactive vascular endothelial cells, causes an increase in portal venous inflow [
20].
The pathophysiology of portal hypertension has also been linked to intrahepatic microvascular thrombosis. While, in the past, cirrhosis was considered a pro-hemorrhagic condition, it is now accepted that cirrhosis is rather characterized by a delicate hemostatic balance [
21]. Parenchymal extinction, which results from the death of the hepatocytes and their replacement with fibrotic tissue following the thrombotic occlusion of intrahepatic veins and sinusoids, is involved in the progression of cirrhosis and in worsening portal hypertension [
22,
23,
24]. Several studies have demonstrated that anticoagulation therapy can reduce hepatic fibrosis and portal hypertension, and delay hepatic decompensation [
25,
26].
3. Gut–Liver Axis Composition and Function
Everything that connects the intestine to the liver contributes to the realization of the gut–liver axis.
The intestinal barrier is the most exposed part to the external environment; it is composed of the mucus layer, produced by intestinal goblet cells [
27], the enterocytes connected by intercellular tight junctional complexes [
28], Paneth cells, the gut-associated lymphoid tissue (GALT) [
29,
30,
31], and the gut vascular barrier [
32]. The gut microbiome resides on top of the intestinal barrier, in the intestinal lumen and the outer mucus layer, along with many substances that serve as host defense and regulate the gut ecosystem, such as antimicrobial peptides (AMPs), IgA, and bile acids [
33,
34,
35,
36,
37].
Under normal conditions, a limited amount of gut microbiome-associated molecular patterns (MAMPs) and pathogen-associated molecular patterns (PAMPs), which include lipopolysaccharide (LPS) and other components of the bacterial cell membrane, flagellin, and bacterial DNA [
38], can cross the epithelial barrier. These molecules activate pattern-recognition receptors (PRRs) on antigen-presenting cells and on B and T cells located in the GALT and the mesenteric lymph nodes (MLNs) [
29,
30,
31,
39], which is crucial for modeling the immune system and avoiding a systemic immune response [
40].
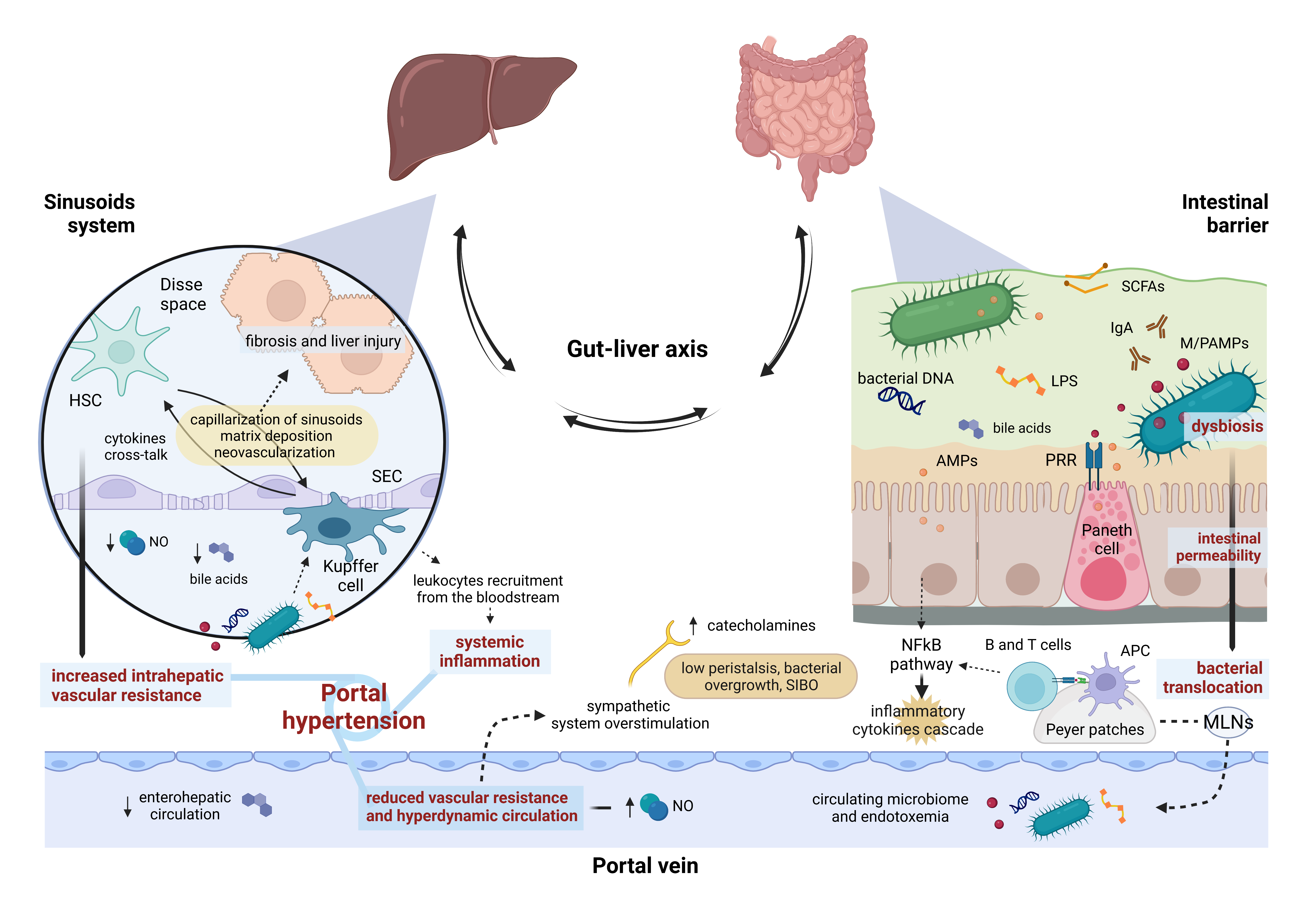
On the inner side of the gut–liver axis, the hepatic sinusoids act as the final filter of the substances collected by the splanchnic vessels. This functional unit is composed of fenestrated sinusoidal endothelial cells (SECs), resident macrophages named Kupffer cells, and hepatic stellate cells (HSCs); the latter are located in the space of Disse, between the endothelium and the hepatocytes, and are involved in tissue repair and fibrogenesis [
41,
42].
In summary, the gut–liver axis is an extremely dynamic system, regulated by several host cytokines, vasoactive mediators, and microbial metabolites, in a constant balance between pro-inflammatory and tolerance factors [
43,
44,
45]. The disruption of its homeostasis participates in the development of portal hypertension, which leads to the dysfunction of the gut–liver axis at several points, not only causing liver damage and systemic inflammation, but also worsening liver hemodynamics in a vicious cycle (
Figure 1).
Figure 1. Gut–liver axis and portal hypertension. The intestinal barrier and the hepatic sinusoid system represent the two hinges of the firewall involved in containing bacterial translocation within the gut–liver axis. Impairment of continuous, bi-univocal communication between all these elements at several points gives rise to a vicious cycle leading to portal hypertension. AMPs, antimicrobial peptides; APC, antigen-presenting cell; HSC, hepatic stellate cell; LPS, lipopolysaccharide; M/PAMPs, microbiome/pathogen-associated molecular patterns; MLNs, mesenteric lymph nodes; NO, nitric oxide; PRR, pattern-recognition receptor; SCFAs, Short-chain fatty acids; SEC, sinusoidal endothelial cell; SIBO, small intestinal bacterial overgrowth.
4. Gut–Liver Axis Impairment and Portal Hypertension: A Two-Way Street
The gut microbiome shows qualitative and quantitative alterations in cirrhotic patients, and portal hypertension plays a central role in this process through intestinal mucosal congestion and atrophy, which reduce gastric acid production and peristalsis, impairing bacterial clearance [
10,
40,
46]. This mechanism results in a reduced ratio between autochthonous and potentially pathogenic taxa [
47], with a decrease in
Lactobacillus, Bifidobacterium,
Ruminococcaceae,
Lachnospiraceae,
Clostridium cluster IV, and
Bacteroides, and an increase in
Streptococcus,
Veillonella,
Fusobacterium, Enterococcaceae, and
Proteobacteria (in particular
Enterobacteriaceae) [
13,
48,
49]. Small intestinal bacterial overgrowth (SIBO) is also a frequent finding in patients with cirrhosis, which can be explained by the impaired intestinal motility associated with the high sympathetic tone in portal hypertension [
50,
51].
Bacterial translocation has been recognized as a key pathological mechanism triggering the onset and the progression of portal hypertension. In cirrhotic patients, the abnormal bacterial translocation overcomes MLNs’ defense capacity, consequently engaging the sinusoid system [
19,
52,
53,
54,
55,
56]. Kupffer cells are overstimulated in producing pro-inflammatory mediators, such as interleukin 6 (IL-6), tumor necrosis factor-alpha (TNF-alpha), and chemokines, through a series of pathways, including toll-like receptor 4 (TLR-4), Myeloid differentiation primary response 88 (MyD88), and nuclear factor kappa B (NFκB), in a cross-talk with SECs and HSCs, which acquire an activated, fibrinogenic phenotype [
10,
42,
45,
57,
58,
59,
60]. All of this results in a series of maladaptive consequences: capillarization of liver sinusoids, extracellular matrix deposition and fibrosis, liver damage, and neovascularization [
61,
62]. Nevertheless, the inflammatory response extends beyond the liver; inflammatory mediators overflow into the systemic circulation, causing the recruitment of leukocytes from the bloodstream [
63,
64] and the release of vasoactive mediators. Among others [
65,
66], NO, produced by endothelial and inducible NO synthases (eNOS and iNOS), plays a key role in steering the hemodynamic changes in liver disease. While NO is reduced in the intrahepatic microcirculation, causing vascular hypertonus and increasing microvascular resistance, in the splanchnic and systemic bed, both iNOS and eNOS are upregulated, resulting in arterial vasodilation, reduced vascular resistance, and hyperdynamic circulation [
40,
67,
68,
69].
Therefore, gut–liver axis disruption plays a crucial role in the development and progression of portal hypertension [
70]. Although it is difficult to determine whether the chicken or the egg comes first and, in particular, at what point of liver disease, growing attention has been paid to identifying the mechanisms through which the gut microbiome can modulate portal hypertension.
5. Influence of the Gut Microbiome on Portal Hypertension
Recently, various studies have suggested a strong interplay between the gut microbiota and the development and progression of portal hypertension (
Table 1) [
13,
14].
Table 1. Features of the gut microbiome associated with portal hypertension in animal models and human studies.
A recent study comparing conventional and germ-free mice showed that the presence of the gut microbiota stimulates the proliferation of vessels and lymphatic collectors in the intestinal wall, which depends, at least in part, on the production by Paneth cells of Angiogenin-4 (Ang-4), a ribonuclease with angiogenic and antimicrobial properties [
77,
78]. This was paralleled by the increase in portal pressure, outlining the hypothesis that gut microbiota may per se drive portal hypertension, irrespective of bacterial translocation, systemic inflammation, and the development of ACLD [
79].
Microbial metabolites represent an additional pathophysiological link between portal hypertension and the gut–liver axis. Hydrogen sulfite (H2S) [
14] is produced by sulfur-reducing gut bacteria (i.e.,
Bilophila and
Desulfovibrio genera, both belonging to the
Proteobacteria phylum) and by the host via H2S-catalyzing enzymes variably expressed in many organs [
80]. H2S induces vasodilation when interacting with endothelial and smooth muscle cells, and suppresses the contraction of HSCs in experimental cirrhosis [
81,
82]. Furthermore, it reduces gastrointestinal motility, favoring bacterial overgrowth and the development of SIBO [
83].
Short-chain fatty acids (SCFAs) (acetic acid, propionic acid, butyric acid, isobutyric acid, valeric acid, and isovaleric acid) result from the gut microbiome fermentation of non-absorbable carbohydrates; they regulate the function of the intestinal barrier both directly, providing energy to enterocytes, and indirectly, exerting anti-inflammatory effects on the innate and adaptive immune system [
84,
85,
86]. SCFAs have been found in the portal and peripheral blood, participating in several processes, including the modulation of liver hemodynamics [
87]. A study enrolling 62 patients with cirrhosis showed how the level of circulating SCFAs was inversely associated with the severity of liver disease and the model for end-stage liver disease (MELD) score; above them, butyric acid was inversely correlated with the HVPG values, inflammatory markers, such as TNF-alpha and IL-6, and NO in hepatic, peripheral, and portal blood [
88]. Nevertheless, SCFAs were related to hemodynamic changes, not only at a portal level, but also at a systemic level, being directly correlated with systemic vascular resistance and inversely correlated with the cardiac index.
Bile acids are key protagonists of intestinal functions and act as signaling molecules, regulating several metabolic and physiological processes. Bile acids have anti-microbial and immune-modulating properties in the gut [
89,
90] while participating in the regulation of intrahepatic vascular resistance by interacting with the sinusoid system via farnesoid x receptor (FXR) signaling [
91,
92]. In advanced cirrhosis, both primary and particularly secondary bile acid production is reduced [
93], thus contributing to dysbiosis, SIBO, and bacterial translocation [
13], as well as altered sinusoidal vasoregulation, consequently affecting the progression and the severity of portal hypertension.
Antimicrobial peptides (AMPs) are a wide and diverse group of small proteins implied in the host–microbiome interplay [
94,
95,
96]. Defensins, cathelicidins, and lectins are the most common AMPs in the gut, mainly derived from Paneth cells and enterocytes; they operate in a complex and synergistic dynamic in the regulation of the gut microbiome, both directly by damaging microbes and indirectly by interacting with the host intracellular signaling pathways and stimulating the immune response [
95]. Many intestinal bacterial strains also produce specific AMPs, i.e., the bacteriocins, involved in the mechanisms of bacterial competition and communication, as well as biodiversity and environmental niche formation [
97,
98,
99]. There is some evidence of the relevance of the AMPs network in liver disease. In mice models of ethanol-induced liver injury, a deficiency of the regenerating islet-derived 3 beta and gamma (REG3B and REG3G), two gut lectins with bactericidal properties against Gram-negative bacteria, was associated with an increase in mucosa-associated bacteria and in bacterial translocation, together with worsening disease progression [
100]. Of interest, in experimental cirrhosis, increased bacterial translocation was associated with a depletion of Paneth cells and a reduced expression of AMPs [
101]; however, in the same study, this association was not observed in mice with acute portal hypertension without cirrhosis. While a considerable amount of data is already available on the gut microbiota composition associated with liver cirrhosis, the detailed analysis of which is beyond the scope here, evidence on microbiota signatures associated with portal hypertension and its severity is still lacking. Some attempts have been made to demonstrate the value of the gut microbiota profile as a noninvasive diagnostic marker of portal hypertension. In particular, the circulating microbiome has been identified as a possible target in this context, reasonably mirroring bacterial translocation from the gut [
49,
102,
103]. A recent study aimed to find microbial signatures of portal hypertension in blood compartments of patients with cirrhosis, particularly in peripheral circulation and hepatic veins [
71]. While there were significant differences in the circulating microbial composition compared with controls and in association with MELD or biomarkers of inflammation, no predictive value regarding portal hypertension severity could be demonstrated. It has also recently been shown that specific components of the microbiome in the peripheral blood flow at baseline can predict the reduction in HVPG after direct-acting antiviral (DAA) therapy in HCV-related cirrhosis [
72]. However, the study enrolled only 32 patients, including people with HIV and HCV coinfection, making it difficult to draw strong conclusions. Another study analyzed the gut microbiome of 12 inpatients with esophageal and gastric varices compared with 24 healthy controls, showing a higher relative abundance of
Lactobacillales and a reduction in
Clostridium cluster IV and cluster IX [
73]; in this setting, no distinction was made concerning the severity of portal hypertension or in comparison with cirrhotic patients without CSPH.
This entry is adapted from the peer-reviewed paper 10.3390/microbiolres13030038