Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Others
Immunoglobulin A nephropathy (IgAN) is a rare autoimmune disorder and the leading cause of biopsy-reported glomerulonephritis (GN) worldwide. Disease progression is driven by the formation and deposition of immune complexes composed of galactose-deficient IgA1 (Gd-IgA1) and Gd-IgA1 autoantibodies (anti-Gd-IgA1 antibodies) in the glomeruli, where they trigger complement-mediated inflammation that can result in loss of kidney function and end-stage kidney disease (ESKD).
- IgAN
- CD38
- immune system
1. New Strategies for the Management of IgAN
The immune system plays a multifaceted role in initiating and promoting the loss of kidney function seen in patients with IgAN [1,16,17,20]. Growing clinical data support approaches that deplete Gd-IgA1-producing cells or reduce immune complex-mediated inflammation, which hold greater promise than broad-acting immunosuppressive treatments (ISTs) that have historically demonstrated a lack of efficacy or were associated with significant toxicity.
2. Inhibition of Immune Complex-Activated Complement Activity
There is pathologic biochemical and genetic data supporting the pivotal role of the complement system in the pathogenesis and progression of IgAN that is now well established [20]. Accumulating evidence suggests that activation of both the alternative and lectin pathways, leads to glomerular inflammation and injury in IgAN [20,43,45,46,47]. Here, we review several complement inhibitors that are in advanced stages of clinical development.
Iptacopan (LNP023, Novartis, Basel, Switzerland) is an investigational, oral small molecule complement factor B inhibitor of the alternative pathway that is being evaluated in adults with IgAN [48,49]. Results from a Phase 2 clinical trial (NCT03373461) demonstrated a potential for effective and clinically meaningful reduction in proteinuria. The trial randomized 112 patients with IgAN into three dosing arms of iptacopan and a placebo arm. Results showed the highest dose of iptacopan (200 mg, twice daily) can reduce urine protein: creatinine ratio (UPCR) by 40% from baseline to 6 months, compared with placebo. Based on these encouraging data, the Phase 3 APPLAUSE-IgAN trial has been launched and is currently ongoing (NCT04578834) [48,50].
Similarly, narsoplimab (OMS721, Omeros, Seattle, WA, USA), an investigational humanized monoclonal antibody selectively targeting mannan-binding lectin-associated serine protease-2 (MASP-2), is a novel pro-inflammatory protein target and the effector enzyme of the lectin pathway that is being evaluated in patients with IgAN [20,51,52]. Three-year follow-up data from a Phase 2 clinical trial (NCT02682407) in 12 high-risk patients with advanced IgA nephropathy showed a median reduction in proteinuria of 64.4% and long-term improvement or sustained stabilization in eGFR when treated with narsoplimab [51,53]. A Phase 3 trial is currently ongoing (NCT03608033) [20].
Iptacopan and narsoplimab target the alternative and lectin pathways, respectively, leaving the classical complement pathway intact and able to respond to pathogens [54,55].
While complement inhibition has the potential to reduce proteinuria and slowing down eGFR loss, continued production and deposition of immune complexes in the glomeruli may require long-term treatment with complement inhibitors to prevent the progression of kidney disease.
New treatment strategies aim to reduce immune complex formation and subsequent inflammation by targeting sources of Gd-IgA1 and its anti-Gd-IgA1 antibody production.
3. Targeting Cytokines Responsible for B Cell and Plasma Cell Activation and Survival
Under normal conditions, B cells and plasma cells play an important role in producing antibodies that help defend against a multitude of infections. In autoimmune disorders, these same cells can exacerbate or contribute to the disease by producing autoantibodies [15,56]. BAFF and APRIL, cytokines from the tumor necrosis factor family, are known to mediate B cell and plasma cell function and survival [57]. BAFF and APRIL can activate the NF-kB pathway by binding to several cell surface receptors, including transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI), which promotes plasma cell survival and can stimulate IgG antibody production [57,58]. Belimumab (Benlysta, GSK, Brentford, UK), a monoclonal antibody targeting soluble BAFF, is designed to inhibit activation of B cells and plasma cells thought to drive production of pathogenic antibodies in several autoimmune disorders [59]. Belimumab has been approved for systemic lupus erythematosus (SLE) and/or lupus nephritis, and is currently being investigated in addition to rituximab in a Phase 2 trial (NCT03949855) for primary membranous nephropathy (MN), an autoimmune kidney disease with up to 20% chance of progression to ESKD [60,61].
Increased APRIL expression has been observed in patients with IgAN and is correlated with increased expression of Gd-IgA1 antibodies [58]. Targeting APRIL has the potential to limit antibody production in autoimmune-associated plasma cells. This concept is supported by results from the first cohort of a Phase 1/2 study (NCT03945318) evaluating BION-1301 (Chinook Therapeutics, Seattle, WA, USA), an anti-APRIL monoclonal antibody, in up to 40 patients with IgAN showing sustained reduction in levels of Gd-IgA1 antibodies and proteinuria [62]. Similarly, dual targeting of both BAFF and APRIL with atacicept (Vera Therapeutics, Brisbane, CA, USA), a soluble TACI-Ig fusion protein, showed reduction in Gd-IgA1 antibody levels and proteinuria when evaluated in a Phase 2 study (NCT02808429) in 16 patients with IgAN [63]. Atacicept is being evaluated for IgAN in a Phase 2b trial (ORIGIN; NCT04716231) [64]. In addition, a Phase 2 trial with telitacicept (RemeGen, Yantai, China), a soluble TACI-Ig fusion protein, in 44 patients with IgAN also showed the proteinuria reduction (NCT04905212) [65,66]. Anti-APRIL antibody, sibeprenlimab (VIS649, Visterra/Otsuka, Cambridge, MA, USA/Tokyo, Japan), is being studied for safety and efficacy in a Phase 2 clinical trial (NCT04287985) [67,68].
These data support BAFF and APRIL inhibition as a potential treatment for IgAN; however, further studies are required to understand the broader impacts on immunogenicity when altering function of both B cells and plasma cells.
4. Tarpeyo, a Targeted Approach for Immune Cell Depletion in the Small Intestine
Gut-associated lymphoid tissue (GALT), including Peyer’s Patches, which are thought to contain a high concentration of conventional surface IgA1-expressing primed mucosal B cells and plasma cells, may be responsible for the production of Gd-IgA1 in IgAN [69,70]. Although other studies suggest the nasopharynx-associated lymphoid tissue (NALT) or palatine tonsils may also play an important role in Gd-IgA1 production in patients with IgAN [71,72]. Undoubtedly, reducing Gd-IgA1 levels is a promising approach for disease modification [36]. Targeted corticosteroids, such as targeted-release budesonide (Tarpeyo), have been shown to provide effective modulation and reduction in the immune cells within the gut, including long-lived plasma cells and memory B cells [32,69,73]. Tarpeyo is designed to deliver a delayed release formulation of budesonide to the distal ileum, where it locally suppresses immune cell activity, including Gd-IgA1-producing cells and reduce circulating immune complexes that cause downstream inflammation and kidney impairment [69,74,75].
In a Phase 2b study including 150 patients with IgAN and persistent proteinuria despite optimized RAS blockade (NEFIGAN; NCT01738035), Tarpeyo significantly reduced proteinuria levels and stabilized kidney function [76]. Subsequent results from the treatment period of a Phase 3 study (NefIgArd; NCT03643965) evaluating Tarpeyo in 360 patients with IgAN and persistent proteinuria despite optimized RAS blockade showed treatment with Tarpeyo reduced proteinuria by 27% and stabilized eGFR at 9 months compared with placebo, which led to its conditional accelerated approval by the FDA [77,78].
These data suggest that reducing activity of Gd-IgA1-producing immune cells, which would also reduce levels of anti-Gd-IgA1 antibodies and subsequent immune complex formation and deposition, could improve patient outcomes. However, further evidence is needed to confirm that Tarpeyo reduces Gd-IgA1-producing immune cells in Peyer’s Patches.
4. Velcade, Plasma Cell Depletion via Proteasome Inhibition
Additional support for depletion of plasma cells has been observed with VELCADE® (bortezomib, Millennium/Takeda, Cambridge, MA, USA), a proteasome inhibitor that depletes plasma cells and is approved for treatment of MM, has also shown promise in an open-label pilot trial (NCT01103778) which enrolled 8 people with IgAN. Results showed three participants achieved complete remission (proteinuria of <300 mg/day) after treatment for 1 year, suggesting targeting plasma cells through proteasome inhibition could reduce proteinuria in patients with IgAN [79]. However, larger trials are needed to better assess safety and efficacy in patients with IgAN.
5. Felzartamab, Targeted CD38+ Plasma Cell Depletion
The clinical data supporting targeting of Gd-IgA1-producing immune cells in the gut showed improved outcomes for patients but not complete resolution of the disease [77]. Targeting multiple locations and types of autoantibody-producing plasma cells and thereby potentially reducing Gd-IgA1 as well as anti-Gd-IgA1 antibody levels at the same time may contribute to a more robust improvement in patient outcomes.
Felzartamab (MOR202/TJ202, MorphoSys, Planegg, Germany), a fully human immunoglobulin G1 (IgG1) monoclonal antibody designed to target the highly expressed CD38 cell surface antigen on plasma cells, is being evaluated as a potential first-in-class immunotherapy in a Phase 2a clinical trial for patients with IgAN (IGNAZ; NCT05065970) [80]. Binding of felzartamab to CD38+/CD20− plasma cells is thought to induce cell killing through two complementary mechanisms of action (MoA) including antibody-dependent cell-mediated cytotoxicity (ADCC) via natural killer cells and antibody-dependent cell-mediated phagocytosis (ADCP) via macrophages (Figure 1) [16,81,82,83]. Complement-dependent cytotoxicity (CDC) is described to play a role in infusion-related reactions (IRRs), but based on in vitro testing, felzartamab does not trigger CDC or anti-drug antibodies [82,84].
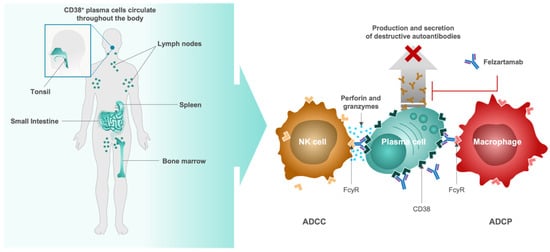
Figure 1. Proposed mechanism of action of felzartamab (MOR202/TJ202) for depleting antibody and auto-antibody-producing CD38+ plasma cells. ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cell mediated phagocytosis; CD, cluster of differentiation; FcγR, Fc-gamma receptor; NK, natural killer.
Preliminary results from a Phase 1b/2a, proof-of-concept trial (M-PLACE, NCT04145440) of felzartamab in 31 patients with anti-phospholipase A2 receptor (PLA2R) antibody-positive MN showed a 46.1% reduction in pathogenic anti-PLA2R autoantibody levels after 1 week in 89% (24/27) of patients with evaluable results. The reduction was sustained, and most patients showed a further increase in reduction over time (12-week treatment). These results support successful and sustained depletion of CD38+ plasma cells with felzartamab [85]. Although further trials are required to collect safety data and address any concerns about therapies that modulate the immune system, the safety profile of felzartamab in the M-PLACE trial was found to be consistent with the proposed MoA, and treatment-emergent adverse events were manageable in patients with MN [85]. Treatment-emergent adverse events (TEAEs) occurred in 26/31 patients and were mostly mild or moderate in severity and the majority resolved. A total of 5 patients experienced treatment-emergent serious adverse events, 2 of which were related to felzartamab (type-I hypersensitivity and grade 3 IRR), and no events resulted in death [86].
Anti-CD38 activity has also been established for felzartamab in multiple myeloma (MM) clinical trials. In a Phase 1/2a clinical trial (NCT01421186) evaluating felzartamab in 91 adults with relapsed/refractory (r/r) MM, felzartamab reduced M-protein levels, supporting systemic depletion of CD38+ plasma cells [82,87]. Limited downregulation of CD38 on MM cells was also observed in patients treated with felzartamab, indicating potential for sustained efficacy [88].
Taken together, the preliminary efficacy and safety data on felzartamab in MN and the proof-of-concept results in r/r MM provide further support for clinical development of anti-CD38 antibody therapies in IgAN and highlight their potential application in other plasma cell-driven autoimmune diseases.
As with any immunomodulatory therapies, there is an increased risk of infection due to downregulation of the natural immune defenses. With therapeutics such as complement inhibitors, there is a potential for disrupting the innate immune system, which is one of the first immunologic responses to pathogenic bacteria [89]. The complement pathway, however, does have redundancies within the three main pathways—classical, lectin and alternative—that allow for therapeutic targeting of this pathway while mitigating risk of infection [54,55]. When depleting B cells with general immunosuppression therapies, such as systemic glucocorticoids, there is potential to disrupt the adaptive immune system, which is responsible for clearing bacteria, viruses, fungi, and parasites [90]. Taking a more targeted approach, as is the case with several of the therapies in development, could reduce the risk of infection and potentially improve the risk/benefit profile in patients treated for IgAN.
This entry is adapted from the peer-reviewed paper 10.3390/jcm11102810
This entry is offline, you can click here to edit this entry!