Retrotransposons, a large and diverse class of transposable elements that are still active in humans, represent a remarkable force of genomic innovation underlying mammalian evolution.
- SINE
- ERV
- LINE-1
- transposable elements
- enhancer
- exaptation
1. Introduction
A large fraction of most eukaryotic genomes is constituted by transposable elements (TEs), interspersed repeats of which the high copy number reflects mobile DNA integration events that occurred countless times throughout evolutionary history [1]. Although they represent a constant challenge for genome stability, TEs have at the same time introduced potentially fruitful changes into genomes both by driving genomic rearrangements (resulting, for example, in gene duplication) and by exaptation of TE-derived sequences [2,3,4]. Since TEs replicate as genomic parasites, eukaryotic host organisms have co-evolved TE silencing systems largely based on the deposition of repressive epigenetic marks, of which the effect on the epigenome accompanied the effect of TE on the genome over the course of evolution [5]. By allowing TE retention, TE silencing mechanisms have provided genomes with large pools of latent functional elements poised for exaptation [6]. While the so-called DNA transposons employ a mechanism directly moving DNA segments from one genomic location to another, the heterogeneous class of TEs referred to as retrotransposons (or retroelements) do so through reverse transcription of an RNA copy of the original element, thereby affecting genome composition through the constant introduction of new DNA material. The peculiar ability of retrotransposon systems to support conversion from RNA to DNA incessantly contributes new sequences, potentially encoding new protein/RNA molecules or providing new cis-regulatory functions that selection can act on to produce genomic and organismal innovations [7,8].
The last 15 years have seen a remarkable spurt of studies exploring the idea that retrotransposon activity affects brain development and function in mammals, through the promotion of somatic mosaicism in the brain [9,10], the generation of novel transcripts and proteins playing diverse roles in neuron biology [11,12], as well as the seeding of cis-regulatory elements, affecting transcription factor-dependent gene regulation, and of boundary elements, participating in three-dimensional (3D) genome architecture [13,14]. The deep involvement of retrotransposons in brain biology also makes them a source of vulnerability and disease [15,16]. All these indications of the pervasive influence of retrotransposons on brain biology consolidate the idea that the evolution of the nervous system, of which the results include the uniquely evolved human brain, has retrotransposons among its driving factors [13,17,18].
2. Retrotransposons and Their Impact on Mammalian Genome Evolution
2.1. Retrotransposons in Mammalian Genomes
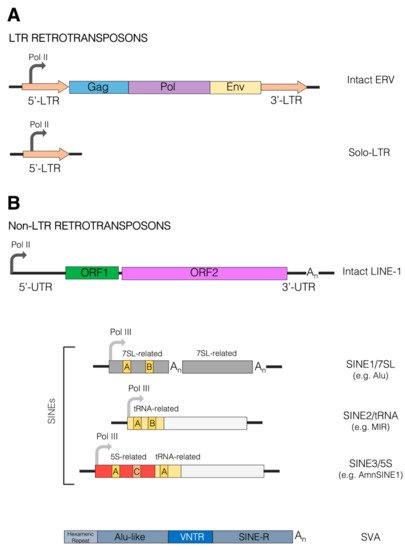
2.2. Retrotransposons as Drivers of Genomic Rearrangements
2.3. Retrotransposon Exaptation as a Source of Genomic Novelties
The exaptation of retrotransposed sequences, consisting in their cooption for a current function out of a hitherto neutral evolution mode, is a well-documented phenomenon [2,74]. In general, large-scale DNA editing of retrotransposons, by simultaneously generating large numbers of mutations, may have accelerated their exaptation during mammalian evolution [81]. In a similar vein, inverted SINE repeats being part of longer RNAs may have promoted RNA editing by adenosine to inosine deamination, thus generating potential novelties in both coding and regulatory sequences [82].
For simplicity, two major exaptation modes can be distinguished. According to the first mode, retrotransposon-derived sequences become physical and functional parts of transcription products, even being eventually translated into protein sequences. The second mode consists in the co-optation of retrotransposon-derived sequences as transcription regulatory elements or 3D genome boundary elements. This exaptation mode, allowing retrotransposon sequences to exert their influence without becoming incorporated into gene products, might have had an even wider influence on genome evolution [83,84].
2.3.1. Retrotransposon-Derived Sequences within Gene Products
As to the first mode of action, there is solid evidence that SINE (in particular, primate-specific Alu) exonization contributes to both untranslated and protein-coding regions of mRNAs [85], as well as portions of long noncoding RNAs [86], to which the embedded Alu can confer new regulatory functions [87]. Alu-derived exons are often the site of alternative splicing, due to the presence in the Alu body of multiple cryptic splice sites [88]. SVA-mediated transduction events, involving alternative mRNA splicing at cryptic splice sites, have been found to promote exon shuffling and thus genomic novelty [89]. At the same time, cells have evolved precise mechanisms to control Alu and the incorporation of other retroelements within mRNA sequences via their cryptic splice sites, as their incorrect presence might induce devastating physiological responses [90]. Moreover, exonic SINE sequences embedded into the 3’ UTR of mRNAs participate in different layers of post-transcriptional gene regulation, which may also involve intermolecular base-pairing with SINE sequences embedded in lncRNAs [43,88,91,92]. Alu SINEs embedded into precursor transcripts were also found to promote the formation of circRNAs [93], a complex family of eukaryotic regulatory transcripts under intense study [94,95]. There is also abundant evidence for TE-derived microRNAs, some of which are potentially involved in human evolution and disease [96,97,98,99,100].
In the case of autonomous retrotransposons, which contain protein-coding sequences in their body, several striking cases of exaptation of retrotransposon-encoded proteins as new host proteins have been documented. A remarkable example is represented by syncytins, an ensemble of Env proteins coded by different ERVs in the genome of various vertebrates, that through a process of convergent evolution led to the development of the placenta in eutherian mammals [55,101]. In fact, the union between maternal and fetal cells to constitute the placental syncytiotrophoblast—the main site of trophic exchanges during pregnancy—is mediated by the fusogenic activity of syncytins, which changed from being mechanisms of viral entry to exerting physiological activity domesticated to serving the host biology [55]. Some syncytins are indeed thought to have a role in other placenta-associated functions, such as the establishment of maternal immune-tolerance against the fetal allograft through their natural immune-suppressive properties, which in ancestral infections likely guaranteed their immune escape [102,103].
2.3.2. Retrotransposons as a Source of cis-Regulatory Sequences
Given the centrality of cis-regulatory elements, and particularly of enhancers, in orchestrating organ-, tissue- and cell type-specific gene expression both during development and in adult organisms [104], it has been argued that the “vast majority of the genetic changes responsible for the evolution of morphology occur at pre-existing cis-regulatory elements” [105], and that TE-mediated cis-regulatory network rewiring has been one of the key mechanisms for the appearance of such changes [6]. In the last 10–15 years, the exaptation of TE-derived sequences (especially retrotransposon-derived) as cis-regulatory elements has been well documented by a rapidly growing body of studies, the majority of which have focused on mammalian genomes, characterized by the overwhelming prevalence, in terms of both amount and activity, of retrotransposons over DNA transposons. Retrotransposon-derived cis-regulatory sequences have been reported to play several roles in gene regulation as promoters, enhancers, silencers and boundary elements [2,83]. In general, due to their own replicative needs, retrotransposons have evolved cis-acting sequences mimicking those of the host, a fact that predisposes them to cis-regulatory activity [76]. Although we are still far from a comprehensive picture of the multiple layers of TE-derived regulatory novelties and their integration with the whole genomic background of mammalian evolution, various cis-regulatory modes of TE exaptation have begun to be clearly portrayed (Figure 2).
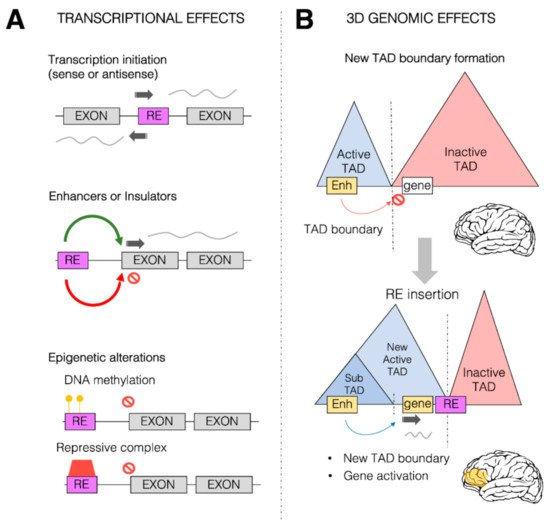
Figure 2. Transcriptional and 3D genome effects of retrotransposons. Retrotransposons (or retroelements, RE) are responsible for a wide range of possible effects on both transcription control and 3D genome organization. (A) Schematic representation of some of the predominant effects of REs on transcription. REs (purple box) can be inserted within the coding region between two exons (gray boxes), providing new transcription start sites (TSS, dark gray arrow) for both sense and antisense transcription. REs can also provide new cis-regulatory sequences (such as enhancers or insulators) which can in turn activate (green arrow) and repress (red arrow) transcription of the associated gene. REs could also alter the epigenetic state of a given gene, leading to its transcriptional repression, by increasing the DNA methylation (yellow circles) within the promoter region of the transcription unit and directly or indirectly recruiting repressive complexes (red box). (B) REs can impact the 3D genome organization of the chromatin within the nuclei. REs (especially Alu elements) are found to be enriched at topologically associating domain (TAD) boundaries. Represented in the Figure is a putative case in which two TADs, one active (blue) and one inactive (red), are separated by a TAD boundary. This boundary limits the action of a brain enhancer region (yellow box) within the active TAD towards a gene (white box) within the inactive TAD, thereby impeding the ectopic brain expression of the gene. As a result of an RE insertion event within the inactive TAD, the 3D genome organization is altered, and a new active TAD is formed due to the boundary effect of the RE. This leads to the spreading of the active TAD over the gene, which allows the brain enhancer region (yellow) to now induce gene activation and therefore its ectopic expression within the brain (yellow area).
First of all, many binding sites for diverse TFs are contributed by retrotransposons, as mainly revealed by genome-wide TF occupancy mapping by chromatin immunoprecipitation coupled with high throughput sequencing (ChIP-seq) [106]. Although some of the TF binding sites carried by TEs are justified by their need to employ host TFs for their own life cycle, others may have been acquired independently through TE propagation mechanisms [34]. Molecular evolution studies have revealed waves of expansion of the TF target repertoire over the course of vertebrate evolution, with TEs majorly contributing to such expansions [107]. TFs tend to bind to TE-provided cognate sites in a species-specific manner, in line with the expansion of different TE subfamilies at different evolutionary timepoints [83]. A striking example of how the evolutionary recruitment of TE-derived TF binding contributed to mammalian evolution is provided by the TE-dependent transformation of the uterine regulatory landscape in the evolution of mammalian pregnancy [108]. An emerging topic that is potentially highly relevant to the exaptation of TE-binding TFs, is that of Krüppel-associated box domain zinc finger proteins (KRAB-ZFPs). The great expansion and diversification in mammals of these TFs has been correlated with the invasion of new endogenous retroelements, which require specialized mechanisms of repression via the binding of specific KRAB-ZPs and subsequent recruitment of the KAP1 corepressor [28]. It is thought that the arms race between KRAB-ZFPs and their target retroelements, facilitated by the evolutionary plasticity conferred on both contenders by the repetitive organization of their genes, favored retroelement domestication, allowing them to develop cis-regulatory functions, to which KRAB-ZFPs have the potential to directly contribute as enhancers or promoter-binding TFs [28,71,109,110].
A second, more complex mode of TE exaptation for cis-regulatory purposes is represented by TE-derived clusters of TF binding sites, exemplified by the contribution of species-specific, composite enhancers to mouse placental development by rodent endogenous retroviruses [111]. In addition, mouse-specific LTRs have been found to carry multiple pluripotency TF-binding sites (specifically, ESRRB-, KLF4- and SOX2-binding motifs) regulating gene expression in a mouse embryonic stem cell (ESC)-specific manner, thereby distinguishing ESCs in mice from ESCs in other species [112]. In a similar vein, recent hominoid-specific LTR and SVA retrotransposons were shown to host enhancers that were active in human naive ESCs and embryonic genome activation [110]. Systematic studies of TEs’ contribution to enhancer function have benefited greatly from high-resolution profiling of the regulatory epigenome, such as the profiling of DNase hypersensitivity, histone H3-lysine 4 mono-methylation (H3K4me1) and histone H3-lysine 27 acetylation (H3K27ac) as typical enhancer chromatin signatures [113] and by the use of a chromatin characterization software such as ChromHMM [114]. A recent comprehensive quantification of the epigenomic status of TEs across many human tissues and cell types revealed that approximately one quarter of the human regulatory epigenome is composed of retrotransposed sequences, with motif-enriched LTRs being particularly favorable substrates for the evolution of new host regulatory elements [115]. In other studies, based on epigenomic profiling, evolutionary novelties in primate gene regulation were similarly found to have TEs as the primary source, with a major contribution from ERV-derived sequences [116,117]. Accordingly, a subset of ERV sequences were found to be significantly enriched in cis-regulatory elements, having a critical role in primate liver gene regulation [117]. A fascinating example of ERV contribution in the shaping of entire regulatory pathways is represented by the interferon (IFN) transcriptional network, a crucial innate antiviral system which also serves as a fundamental effector to initiate and maintain adaptive immunity. Chuong and coauthors showed that ERV insertions had a central role in its evolution and amplification, accounting for the independent dissemination of a wide number of IFN-inducible enhancers in many mammalian genomes, which are required for the correct functioning of different immune responses [118]. A similar scenario is found for p53 tumor suppressor factor, of which the genomic binding sites in humans overlap in more than one-third of cases with ERV elements [119]. Of note, these binding sites are primate-specific and not present in other mammals, further demonstrating that TEs are able to shape important regulatory networks in a species-specific manner. An intriguing observation, consistent with the previous ones, is that of the pervasive function of an ape-specific class of ERV-derived LTRs, LTR5HS, as early embryonic enhancers, regulating hundreds of human genes [120], and the strong contribution of ERV and L1 retrotransposon families to species-specific differences in enhancer activity between chimpanzee and human cranial neural crest cells [83,121]. Epigenome profiling also allowed researchers to distinguish between older retrotransposon copies displaying most of the features of de facto enhancers and younger copies that seem instead to be configured as proto-enhancers, serving as a repertoire for the de novo evolutionary birth of enhancers [122]. Despite the scarcity of studies, an intriguing retrotransposon feature favoring their exaptation as enhancers is their intrinsic capability of generating functional non-protein-coding RNAs (ncRNAs) that could overlap with the so-called enhancer RNAs (eRNAs) [123], thereby raising the possibility that many eRNAs could be generated through TE-derived ncRNAs.
2.3.3. Involvement of Retrotransposons in Three-Dimensional Genome Architecture
Chromosome contacts within the nuclear space, recently revealed at unprecedented resolution by HiC and complementary approaches [124], exert a wide and still largely unexplored influence on gene regulation by demarcating regulatory districts in a highly dynamic way. At a large scale within nuclei, chromosomes segregate into regions of preferential long-range interactions that form two mutually excluded types of chromatin, referred to as “A” and “B” compartments [125], the formation of which has been recently linked to homotypic clustering of L1 and B1/Alu, respectively [126]. At a scale of tens to hundreds of kilobases, chromosomes fold into domains with preferential intradomain interactions known as topologically associating domains (TADs), which harbor the potential to influence enhancer function and thus gene regulatory networks [127,128,129,130,131,132]. TAD demarcation is achieved by specific regions called TAD boundaries, which are enriched for the occupancy of CCCTC-binding factor (CTCF), a zinc finger DNA binding protein also known to mediate the formation of chromatin loops [133]. SINE retrotransposons have also been found to be enriched at TAD boundaries [134,135]. Curiously, in rodents (but not in humans) B2 SINE retrotransposons have been shown to carry CTCF binding motifs, and therefore rodent B2 SINEs can contribute to clustered CTCF sites at TAD boundaries, thus helping in the maintenance of genome organization [136]. However, the rapid expansion of rodent SINEs might provide excessive CTCF sites throughout the genome, therefore critically increasing the possibility of genome mis-folding due to the creation of aberrant CTCF sites. In this context, a complex formed by CHD4, ADNP and HP1 chromatin proteins (ChAHP complex) has been shown to play a role in the maintenance of evolutionarily conserved spatial chromatin organization via the buffering of novel CTCF binding sites that emerge through SINE expansion [137]. Moreover, SINE and other retrotransposons have been proposed to participate in the establishment of species-specific chromatin loops by introducing novel binding sites for architectural proteins, including CTCF [138]. CTCF might also participate, together with other proteins, in the DNA methylation and histone modification boundary activity recently attributed to currently active copies of mouse B2 SINEs, which might be involved in the epigenomic and phenotypic diversification of mouse species [139].
The contribution of retrotransposons to chromatin regulatory domains is not limited to providing CTCF binding clusters. MIR retrotransposons, for example, have been shown to provide regulatory sequences, functioning as insulators in the human genome independently from CTCF [140]. The presence of binding sites for the multi-subunit DNA binding protein TFIIIC is a distinguishing feature of SINEs, and TFIIIC bound to Alu elements has been shown to influence gene regulation through its chromatin looping and histone acetylation capacities [141,142]. In the case of SINEs exapted as enhancers or TAD boundaries, their regulatory function might even take advantage of their Pol III-dependent transcription, which was recently demonstrated to occur with a marked cell-type specificity [123,143,144]. Retrotransposon transcription has also been shown to be required for the cell type- and species-specific chromatin architecture remodeling properties recently attributed to the primate-specific HERV-H TE family of LTR retrotransposons [145].
This entry is adapted from the peer-reviewed paper 10.3390/life11050376