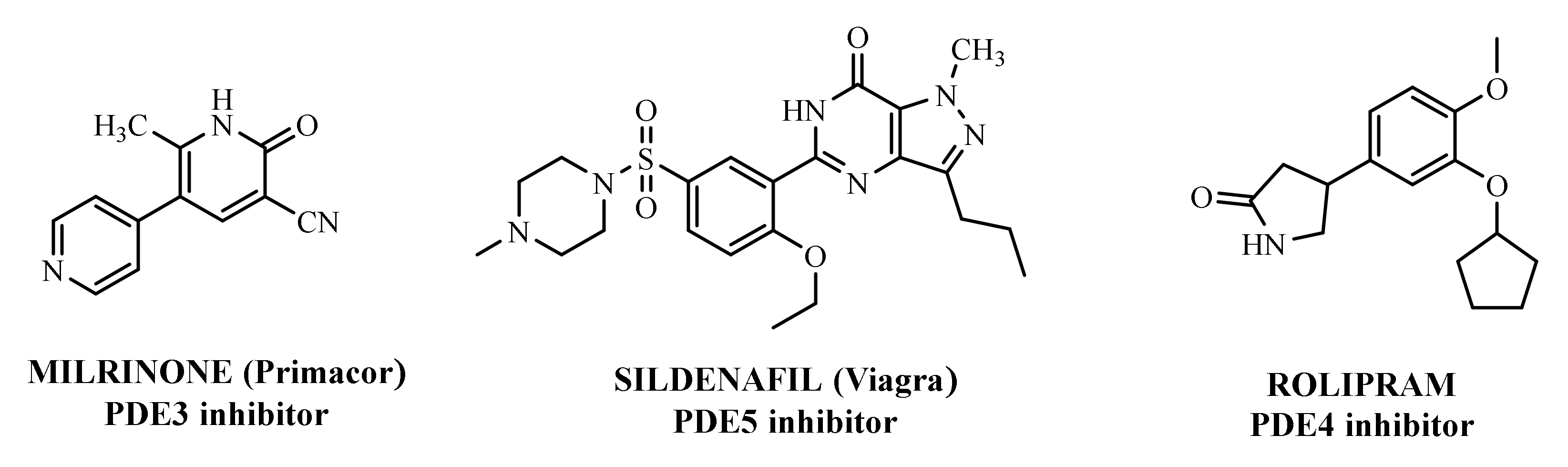
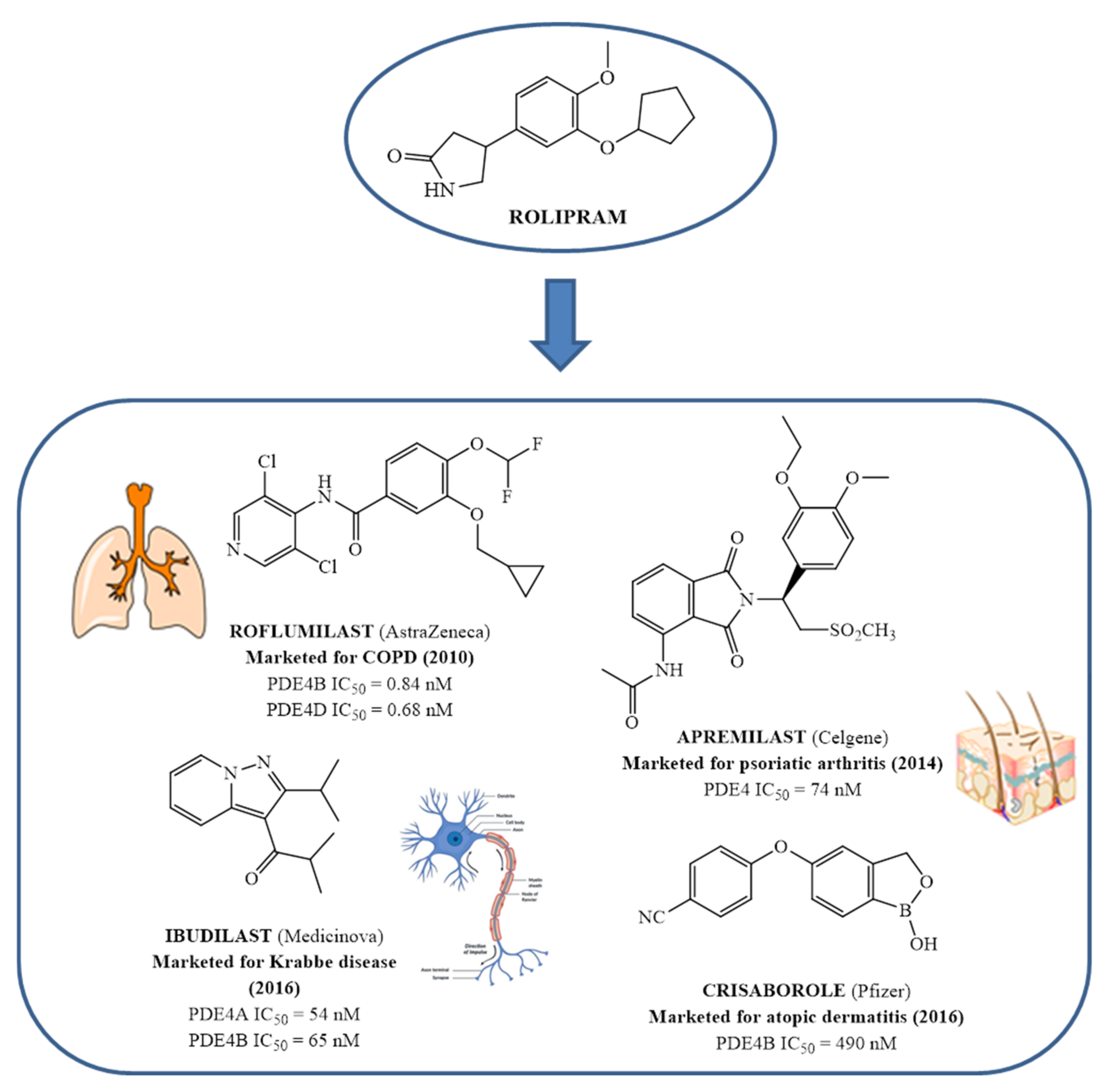
Since the early 1980s, phosphodiesterase 4 (PDE4) has been an attractive target for the treatment of inflammation-based diseases. Several scientific advancements, by both academia and pharmaceutical companies, have enabled the identification of many synthetic ligands for this target, along with the acquisition of precise information on biological requirements and linked therapeutic opportunities. The transition from pre-clinical to clinical phase was not easy for the majority of these compounds, mainly due to their significant side effects, and it took almost thirty years for a PDE4 inhibitor to become a drug i.e., Roflumilast, used in the clinics for the treatment of chronic obstructive pulmonary disease. Three additional compounds have reached the market: Crisaborole for atopic dermatitis, Apremilast for psoriatic arthritis and Ibudilast for Krabbe disease.
- phosphodiesterase 4
- PDE4 inhibitors
- clinical trials
1. Introduction
2. Overview of PDE4
3. Inflammation and PDE4
4. From In Vitro and Preclinical Profile of The First PDE 4 Inhibitors to Approved Drugs
This entry is adapted from the peer-reviewed paper 10.3390/molecules27154964
References
- Sutherland, E.W.; Rall, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091.
- Newton, A.C.; Bootman, M.D.; Scott, J.D. Second messangers. Cold Spring Harb. Perspect. Biol. 2016, 8, a005926.
- Raker, V.K.; Becker, C.; Steinbrink, K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front. Immunol. 2016, 7, 123.
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers. 2014, 6, 436–458.
- Lorigo, M.; Oliveira, N.; Cairrao, E. PDE-mediated cyclic nucleotide compartmentation in vascular smooth muscle cells: From basic to a clinical perspective. J. Cardiovasc. Dev. Dis. 2022, 9, 4.
- Hasegawa, G.R. Milrinone, a new agent for the treatment of congestive heart failure. Clin. Pharm. 1986, 5, 201–205.
- Hilleman, D.E.; Forbes, W.P. Role of milrinone in the management of congestive heart failure. DICP 1989, 23, 357–362.
- Varriale, P.; Ramaprasad, S. Short-term intravenous milrinone for severe congestive heart failure: The good, bad, and not so good. Pharmacotherapy 1997, 17, 371–374.
- Lewis, T.C.; Aberle, C.; Altshuler, D.; Piper, G.L.; Papadopoulos, J. Comparative effectiveness and safety between Milrinone or Dobutamine as initial inotrope therapy in cardiogenic shock. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 130–138.
- Little, W.N.; Park, G.T.; Patton, H.M. Sildenafil in the treatment of erectile dysfunction. N. Engl. J. Med. 1998, 339, 700.
- Goldstein, I.; Lue, T.F.; Padma-Nathan, H.; Rosen, R.C.; Steers, W.D.; Wicker, P.A. Oral sildenafil in the treatment of erectile dysfunction. Sildenafil study group. N. Engl. J. Med. 1998, 338, 1397–1404.
- Cartledge, J.; Eardley, I. Sildenafil. Expert Opin. Pharmacother. 1999, 1, 137–147.
- Antoniu, S.A. Sildenafil citrate for the treatment of pulmonary arterial hypertension. Expert Opin. Pharmacother. 2006, 7, 825–828.
- Ahmed, W.S.; Geethakumari, A.M.; Biswas, K.H. Phosphodiesterase 5 (PDE5): Structure-function regulation and therapeutic applications of inhibitors. Biomed. Pharmacother. 2021, 134, 111128.
- Dal Piaz, V.; Giovannoni, M.P. Phosphodiesterase 4 inhibitors, structurally unrelated to rolipram, as promising agents for the treatment of asthma and other pathologies. Eur. J. Med. Chem. 2000, 35, 463–480.
- Li, H.; Zuo, J.; Tang, W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front. Pharmacol. 2018, 9, 1048.
- Salari-Sharif, P.; Abdollahi, M. Phosphodiesterase 4 inhibitors in inflammatory bowel disease: A comprehensive review. Curr. Pharm. Des. 2010, 16, 3661–3667.
- Richter, W.; Menniti, F.S.; Zhang, H.-T.; Conti, M. PDE4 as a target for cognition enhancement. Expert Opin. Ther. Targets 2013, 17, 1011–1027.
- Wu, C.; Rajagopalan, S. Phosphodiesterase-4 inhibition as a therapeutic strategy for metabolic disorders. Obes. Rev. 2016, 17, 429–441.
- Zebda, R.; Paller, A.S. Phosphodiesterase 4 inhibitors. J. Am. Acad. Dermatol. 2018, 78, S43–S52.
- Mokry, J.; Urbanova, A.; Kertys, M.; Mokra, D. Inhibitors of phosphodiesterases in the treatment of cough. Respir. Physiol. Neurobiol. 2018, 257, 107–114.
- Luo, J.; Yang, L.; Yang, J.; Yang, D.; Liu, B.-C.; Liu, D.; Liang, B.M.; Liu, C.-T. Efficacy and safety of phosphodiesterase 4 inhibitors in patients with asthma: A systematic review and meta-analysis. Respirology 2018, 23, 467–477.
- Bhata, A.; Ray, B.; Mahalakshmi, A.M.; Tuladhar, S.; Nandakumar, D.N.; Srinivasan, M.; Essa, M.M.; Chidambaram, S.B.; Guillemin, G.J.; Sakharkar, M.K. Phosphodiesterase-4 enzyme as a therapeutic target in neurological disorders. Pharmacol. Res. 2020, 160, 105078.
- Phillips, J.E. Inhaled phosphodiesterase 4 (PDE4) inhibitors for inflammatory respiratory diseases. Front. Pharmacol. 2020, 11, 259.
- Peng, T.; Qi, B.; He, J.; Ke, H.; Shi, J. Advances in the development of phosphodiesterase-4 inhibitors. J. Med. Chem. 2020, 63, 10594–10617.
- Liu, Z.; Liu, M.; Cao, Z.; Qiu, P.; Song, G. Phosphodiesterase-4 inhibitors: A review of current developments (2013–2021). Expert Opin. Ther. Pat. 2022, 32, 261–278.
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: New target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398.
- Soderling, S.H.; Beavo, J.A. Regulation of cAMP and cGMP signaling: New phosphodiesterases and new functions. Curr. Opin. Cell Biol. 2000, 12, 174–179.
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isoenzyme as targets of the intracellular signaling network: Benefits of PDE inhibitors in various diseases and perspective for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305.
- Eskandari, N.; Mirmosayyeb, O.; Bordbari, G.; Bastan, R.; Yousefi, Z.; Andalib, A. A short review on structure and role of cyclic-3′,5′-adenosine monophosphate-specific phosphodiesterase 4 as a treatment tool. J. Pharm. Pract. Res. 2015, 4, 175–181.
- Peng, T.; Gong, J.; Jin, Y.; Zhou, Y.; Tong, R.; Wei, X.; Bai, L.; Shi, J. Inhibitors of phosphodiesterase as cancer therapeutics. Eur. J. Med. Chem. 2018, 150, 742–756.
- Sakkas, I.L.; Mavropoulos, A.; Bogdanos, P.D. Phosphodiesterase 4 inhibitors in immune-mediated diseases: Mode of action, clinical applications, current and future perspectives. Curr. Med. Chem. 2017, 24, 3054–3067.
- Card, G.L.; England, B.P.; Suzuki, Y.; Fong, D.; Powell, B.; Lee, B.; Luu, C.; Tabrizizad, M.; Gillette, S.; Ibrahim, P.N.; et al. Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure. 2004, 12, 2233–2247.
- Biagini, P.; Biancalani, C.; Graziano, A.; Cesari, N.; Giovannoni, M.P.; Cilibrizzi, A.; Dal Piaz, V.; Vergelli, C.; Crocetti, L.; Del Canale, M.; et al. Functionalized pyrazoles and pyrazolo pyridazinones: Synthesis and evaluation of their phosphodiesterase 4 inhibitory activity. Bioorg. Med. Chem. 2010, 18, 3506–3517.
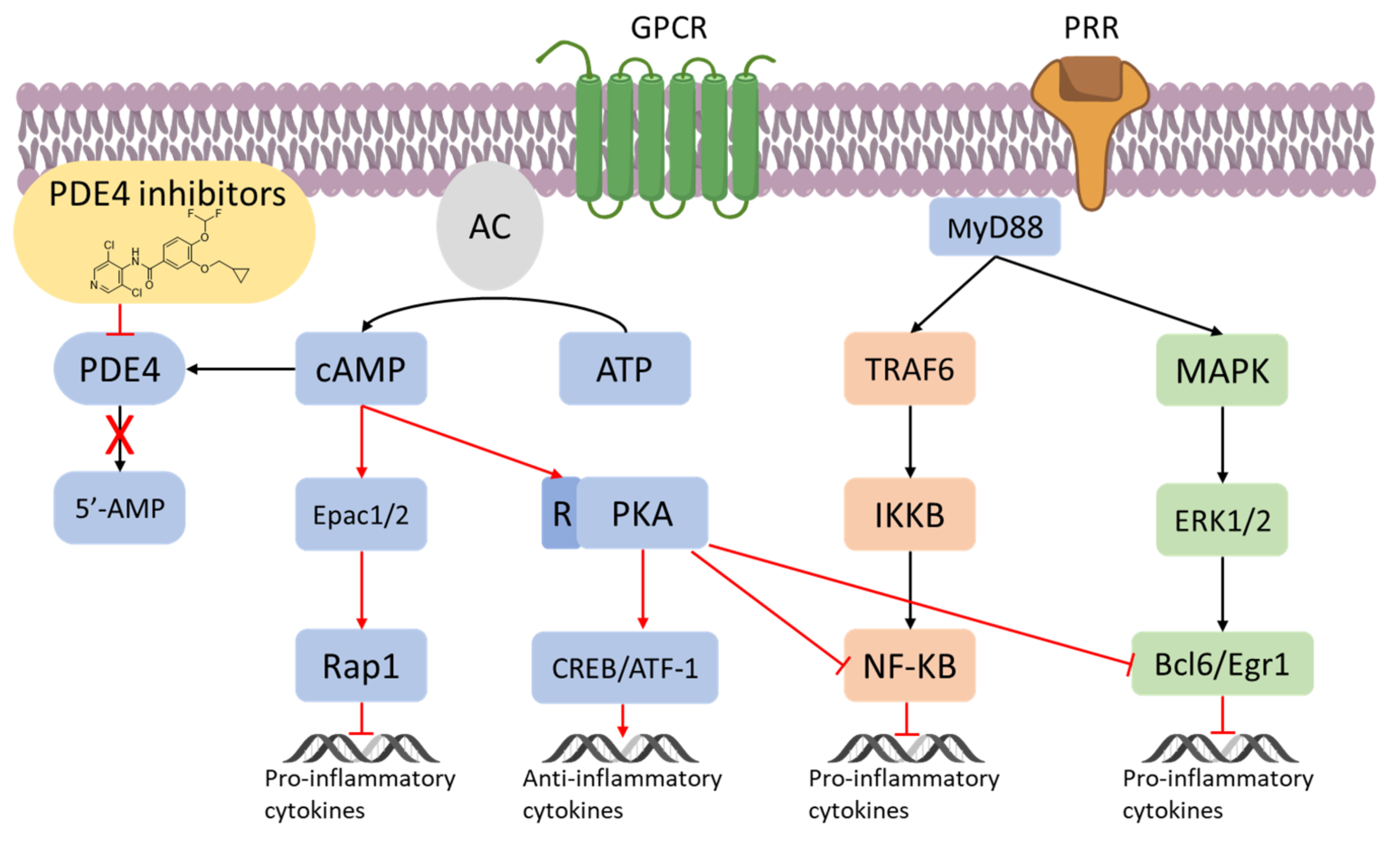
- Conti, M.; Richter, W.; Mehats, C.; Livera, G.; Park, J.-Y.; Jin, C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J. Biol. Chem. 2003, 278, 5493–5496.
- Houslay, M.D.; Milligan, G. Tailoring cAMP-signaling responses through isoform multiplicity. Trends Biochem. Sci. 1997, 22, 217–224.
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-kappaB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596.
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561.
- Schafer, P.H.; Parton, A.; Capone, L.; Cedzik, D.; Brady, H.; Evans, J.F.; Man, H.W.; Muller, G.W.; Stirling, D.I.; Chopra, R. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell. Signal. 2014, 26, 2016–2029.
- Ollivier, V.; Parry, G.C.; Cobb, R.R.; de Prost, D.; Mackman, N. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J. Biol. Chem. 1996, 271, 20828–20835.
- Gobejishvili, L.; Barve, S.; Joshi-Barve, S.; McClain, C. Enhanced PDE4B expression augments LPS-inducible TNF expression in ethanol-primed monocytes: Relevance to alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G718–G724.
- Eigler, A.; Siegmund, B.; Emmerich, U.; Baumann, K.H.; Hartmann, G.; Endres, S. Anti-inflammatory activities of cAMP-elevating agents: Enhancement of IL-10 synthesis and concurrent suppression of TNF production. J. Leukoc. Biol. 1998, 63, 101–107.
- Essayan, D.M.; Huang, S.K.; Kagey-Sobotka, A.; Lichtenstein, L.M. Differential efficacy of lymphocyte- and monocyte-selective pretreatment with a type 4 phosphodiesterase inhibitor on antigen-driven proliferation and cytokine gene expression. J. Allergy Clin. Immunol. 1997, 99, 28–37.
- Bopp, T.; Becker, C.; Klein, M.; Klein-Hessling, S.; Palmetshofer, A.; Serfling, E.; Heib, V.; Becker, M.; Kubach, J.; Schmitt, S.; et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 2007, 204, 1303–1310.
- Bopp, T.; Dehzad, N.; Reuter, S.; Klein, M.; Ullrich, N.; Stassen, M.; Schild, H.; Buhl, R.; Schmitt, E.; Taube, C. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J. Immunol. 2009, 182, 4017.
- Houslay, M.D.; Schafer, P.; Zhang, K.Y. Keynote review: Phosphodiesterase-4 as a therapeutic target. Drug Disc. Today. 2005, 10, 1503–1519.
- Jones, N.A.; Boswell-Smith, V.; Lever, R.; Page, C.P. The effect of selective phosphodiesterase isoenzyme inhibition on neutrophil function in vitro. Pulm. Pharmacol. Ther. 2005, 18, 93–101.
- Schmiechen, R.; Horowski, R.; Palenschat, D.; Paschelke, G.; Wachtel, H.; Kehr, W. 4-(Polyalkoxyphenyl)-2-pyrrolidinones. U.S. Pat. 4,012,495 (to Sobering, A.G.), 15 March 1977. Available online: https://portal.unifiedpatents.com/patents/patent/US-4012495-A (accessed on 13 July 2022).
- Crossland, J. Rolipram. Drugs Future 1988, 13, 38–41.
- Parkes, J.D.; Thompson, C.; Brennan, L.; Gajraj, N.; Howcroft, B.; Ruiz, J. Rolipram in Parkinson’s disease. Adv. Neurol. 1984, 40, 563–565.
- Schneider, H.H.; Schmiechen, R.; Brezinski, M.; Seidler, J. Stereospecific binding of the antidepressant rolipram to brain protein structures. Eur. J. Pharmacol. 1986, 127, 105–115.
- Schudt, C.; Tenor, H.; Hatzelmann, A. PDE isoenzymes as targets for anti-asthma drugs. Eur. Respir. J. 1995, 8, 1179–1183.
- Iona, S.; Cuomo, M.; Bushnik, T.; Naro, F.; Sette, C.; Hess, M.; Shelton, E.R.; Conti, M. Characterization of the rolipram-sensitive, cyclic AMP-specific phosphodiesterases: Identification and differential expression of immunologically distinct forms in the rat brain. Mol. Pharmacol. 1998, 53, 23–32.
- Hartmann, G.; Bidlingmaier, C.; Siegmund, B.; Albrich, S.; Schulze, J.; Tschoep, K.; Eigler, A.; Lehr, H.A.; Endres, S. Specific type IV phosphodiesterase inhibitor rolipram mitigates experimental colitis in mice. J. Pharmacol. Exp. Ther. 2000, 292, 22–30.
- Gobejishvili, L.; Rodriguez, W.E.; Bauer, P.; Wang, Y.; Soni, C.; Lydic, T.; Barve, S.; McClain, C.; Maldonado, C. Novel liposomal rolipram formulation for clinical application to reduce emesis. Drug Des. Devel. Ther. 2022, 16, 1301–1309.
- Maher, A.; El Sayed, N.; Nafea, H.; Gad, M. Rolipram rescues memory consolidation deficits caused by sleep deprivation: Implication of the cAMP/PKA and cAMP/Epac pathways. CNS Neurol. Disord. Drug Targets. 2022, 21, 631–639.
- Brideau, C.; Van Staden, C.; Styhler, A.; Rodger, I.W.; Chan, C.C. The effects of phosphodiesterase type 4 inhibitors on tumour necrosis factor-alpha and leukotriene B4 in a novel human whole blood assay. Br. J. Pharmacol. 1999, 126, 979–988.
- McCann, F.E.; Palfreeman, A.C.; Andrews, M.; Perocheau, D.P.; Inglis, J.J.; Schafer, P.; Feldmann, M.; Williams, R.O.; Brennan, F.M. Apremilast, a novel PDE4 inhibitor, inhibits spontaneous production of tumour necrosis factor-alpha from human rheumatoid synovial cells and ameliorates experimental arthritis. Arthritis Res. Ther. 2010, 12, R107.
- Souness, J.E.; Aldous, D.; Sargent, C. Immunosuppressive and anti-inflammatory effects of cyclic AMP phosphodiesterase (PDE) type 4 inhibitors. Immunopharmacology 2000, 47, 127–162.
- Harbinson, P.L.; MacLeod, D.; Hawksworth, R.; O’Toole, S.; Sullivan, P.J.; Heath, P.; Kilfeather, S.; Page, C.P.; Costello, J.; Holgate, S.T.; et al. The effect of a novel orally active selective PDE4 isoenzyme inhibitor (CDP840) on allergen-induced responses in asthmatic subjects. Eur. Respir. J. 1997, 10, 1008–1014.
- Barnette, M.S.; Underwood, D.C. New phosphodiesterase inhibitors as therapeutics for the treatment of chronic lung disease. Curr. Opin. Pulm. Med. 2000, 6, 164–169.
- Wollin, L.; Bundschuh, D.S.; Wohlsen, A.; Marx, D.; Beume, R. Inhibition of airway hyperresponsiveness and pulmonary inflammation by roflumilast and other PDE4 inhibitors. Pulm. Pharmacol. Ther. 2006, 19, 343–352.
- Giembycz, M.A. Phosphodiesterase 4 inhibitors and the treatment of asthma: Where are we now and where do we go from here? Drugs 2000, 59, 193–212.
- Souness, J.E.; Rao, S. Proposal for pharmacologically distinct conformers of PDE4 cyclic AMP phosphodiesterases. Cell Signal. 1997, 9, 227–236.
- Duplantier, A.J.; Biggers, M.S.; Chambers, R.J.; Cheng, J.B.; Cooper, K.; Damon, D.B.; Eggler, J.F.; Kraus, K.G.; Marfat, A.; Masamune, H.; et al. Biarylcarboxylic acids and -amides: Inhibition of phosphodiesterase type IV versus rolipram binding activity and their relationship to emetic behavior in the ferret. J. Med. Chem. 1996, 39, 120–125.
- Paes, D.; Schepers, M.; Rombaut, B.; van den Hove, D.; Vanmierlo, T.; Prickaerts, J. The molecular biology of phosphodiesterase 4 enzymes as pharmacological targets: An interplay of isoforms, conformational states, and inhibitors. Pharmacol. Rev. 2021, 73, 1016–1049.
- Robichaud, A.; Stamatiou, P.B.; Jin, S.L.; Lachance, N.; Macdonald, D.; Laliberte, F.; Liu, S.; Huang, Z.; Conti, M.; Chan, C.C. Deletion of phosphodiesterase 4D in mice shortens alpha(2)- adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J. Clin. Invest. 2002, 110, 1045–1052.
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S.; et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 2010, 28, 63–72.
- Naganuma, K.; Omura, A.; Maekawara, N.; Saitoh, M.; Ohkawa, N.; Kubota, T.; Nagumo, H.; Kodama, T.; Takemura, M.; Ohtsuka, Y.; et al. Discovery of selective PDE4B inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3174–3176.
- Wedzicha, J.A.; Calverley, P.M.A.; Rabe, K.F. Roflumilast: A review of its use in the treatment of COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 81–90.
- Woo, T.E.; Kuzel, P. Crisaborole 2% ointment (eucrisa) for atopic dermatitis. Skin Ther. Lett. 2019, 24, 4–6.
- Dozier, L.; Bartos, G.; Kerdel, F. Apremilast and psoriasis in the real world: A retrospective case series. J. Am. Acad. Dermatol. 2019, 83, 221–222.
- Rolan, P.; Hutchinson, M.; Johnson, K. Ibudilast: A review of its pharmacology, efficacy and safety in respiratory and neurological disease. Expert Opin. Pharmacother. 2009, 10, 2897–2904.