1. Introduction
Starting from the discovery of phosphodiesterases (PDEs) by Sutherland and Rall in 1958 [
1], there has been a continuous and wide interest by the medicinal chemistry community on the modulation of their activity. PDEs catalyse the hydrolysis of the phosphodiester bond of c-AMP and c-GMP affording the corresponding AMP and GMP inactive counterparts. In fact, the inhibition of PDE leads to an increase in cyclic nucleotide levels, which in turn play a prominent role as second messengers, in the regulation of a variety of cell functions, such as secretion, contraction, metabolism and growth [
2,
3,
4,
5].
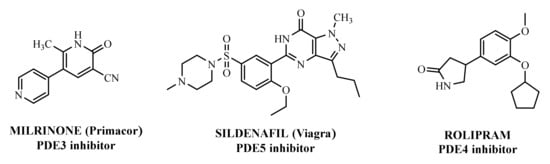
The first important synthetic effort made during the ‘1980s by the pharmaceutical industry in the area of PDE led to the development and marketing by Sanofi-Aventis of the PDE3 inhibitor Milrinone [
6,
7] for the treatment of heart failure (
Figure 1). However, this drug had many drawbacks and was later withdrawn from the market because of serious side effects in the long term. Indeed, chronic administration of Milrinone resulted in reduced survival, showing that chronic elevation of c-AMP in cardiac myocytes is associated with side effects provoking a major risk of fatal arrhythmias [
8]. Milrinone is currently marketed exclusively for hospital use in cases of cardiac shock [
9] and given its power, it is found in numerous clinical trials for pulmonary hypertension, septic shock and other (ClinicalTrials.gov Identifier: NCT04484675: Comparative Study Between Inhaled and Intravenous Milrinone in Patients With Severe Pulmonary Hypertension Undergoing Cardiac Surgery; ClinicalTrials.gov Identifier: NCT05122884: for the treatment/prevention of severe sepsis/septic shock, whose relatively common complication is myocardial dysfunction; ClinicalTrials.gov Identifier: NCT04362527: for the therapy of subarachnoid hemorrhage).
Figure 1. PDEs inhibitors.
Another significant success was the launch of Sildenafil (
Figure 1), the first oral PDE5 inhibitor originally studied for angina, and then approved by FDA and marketed by Pfizer as Viagra
® in 1998 for the treatment of male erectile dysfunction [
10,
11,
12]. About seven years later it was also approved for pulmonary arterial hypertension and classified as an orphan drug by EMEA [
13]. The commercialization of Sildenafil was quickly followed by the marketing of additional PDE5 inhibitors [
14].
This remarkable achievement reinforced the assumption to intensify the investigations also in the field of PDE4 inhibitors, already widely explored as potential candidates for the treatment of chronic inflammatory disorders. In the development of these drugs, the most ambitious goal was to be able to limit the side effects, in order to overcome the difficulties found with PDE3 inhibitors, therefore allowing chronic use.
The interest for PDE4 by both academia and pharmaceutical companies is widely documented by the numerous reviews and patents published over the past twenty years [
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26] and despite the disappointing results obtained in clinical studies with various PDE4 inhibitors, as well as the early termination of trials on compounds under development, very important results have been obtained in this field, as highlighted by the PDE4 inhibitors currently in clinical use.
2. Overview of PDE4
The superfamily of PDEs is currently subclassified into 11 families, namely PDE1-PDE11, which are characterized by different kinetic properties, tissue distribution, responsiveness to endogenous regulators (Ca
2+, calmodulin, c-GMP) and co-factors (Mg
2+, Zn
2+), sensitivity to synthetic inhibitors and, in some cases, substrate specificity (c-AMP or c-GMP) [
27]. Each family is expressed by one or more genes; furthermore, alternative mRNA processing is responsible for the production of multiple splice variants. Thus, until now, more than 50 distinct human PDE proteins have been identified. The generally accepted nomenclature for the different gene products is based on two capital letters indicating the species (HS, Homo sapiens, RT Ratus norvegicus), followed by PDE; then there is an Arabic numeral indicating the family, which is followed, in turn, by a capital letter for the gene (A, B, C, D) and finally by an Arabic numeral for the splice variant [
28,
29].
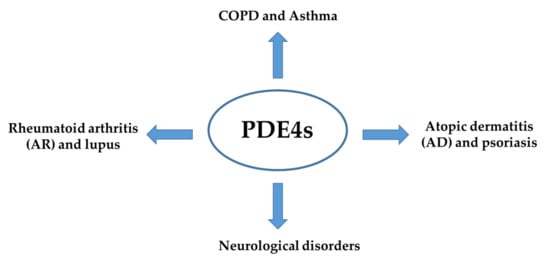
Phosphodiesterase-4 (PDE4) is the most diversified sub-family of phosphodiesterase (PDEs) and is abundantly expressed in a variety of cell types [
30]. These enzymes have been found to mediate several physiological processes, such as brain functions, macrophage and monocyte activation, myocardial contractility, vascular smooth muscle proliferation and neutrophil infiltration, to name a few. Moreover, PDE4 has been reported to participate in the physio-pathogenesis of many inflammatory diseases such as rheumatoid arthritis, chronic obstructive pulmonary disease (COPD) and asthma (
Figure 2). Additionally, PDE4 have shown roles in the progress and development of autoimmune diseases, cardiovascular diseases, and cancers [
31,
32].
Figure 2. PDE4s involvement in the physio-pathogenesis of inflammatory diseases.
Four different subtypes of PDE4 have been identified to date, namely PDE4A, PDE4B, PDE4C and PDE4D. The genes of these subtypes are located on chromosomes 19p13.2, 1p31, 19p13.11, and 5q12, and each of these subtypes can express from 3 to 11 different proteins, resulting in at least 25 different isoforms of PDE4 distributed within different cellular compartments and with different levels of expression. All the PDE4 have been reported to exist in three different forms based on their size i.e., long, short and super-short. The longer version of the protein has two conserved domains, UCR1 (of approximately 60 amino acids) and UCR2 (of approximately 80 amino acids) in the N-terminal region. Differently, the short form has only the UCR2 domain in full, while the super-short version of the protein has a truncated UCR2. At the C-terminus, all the PDE4 have a catalytic domain of 300–350 amino acids. The active site of the enzyme can be divided into three sections as shown by X-ray structures: (i) a pocket that interacts with the phosphate moiety of cAMP, (ii) two pockets that form interactions with small molecules inhibitors, and (iii) a solvated pocket [
33,
34]. Unfortunately, the high conserved sequence identity among the family of PDE4 make the discovery of isoform-selective inhibitors challenging.
The UCRs motifs of the PDE4 are also important for the PDE4 regulation [
35]. PDE4 is regulated by transcriptional regulation (long-term) or post-translational modifications (short-term). When the protein kinase A (PKA) phosphorylates in a conserved PKA phosphorylation site the UCRs, it has been reported to regulate the PDE4 dimerization and catalytic activities [
25]. In long-term regulation of PDE4, c-AMP concentration is increased and the activation of adenylyl cyclase (AC) by hormone mediate stimulation is assisted as a result of increased gene expression. Differently, in the short-term regulation, the activation of PKA is determined by the increased concentration of c-AMP levels. In turn, PKA phosphorylates specific serine residues in UCR1 of PDE4, producing a rapid increase in its activity. However, PDE4 activity is also regulated by other proteins such as Src family tyrosine protein kinases, arrestin and the receptor for activated C kinase 1 (RACK1) [
35].
3. Inflammation and PDE4
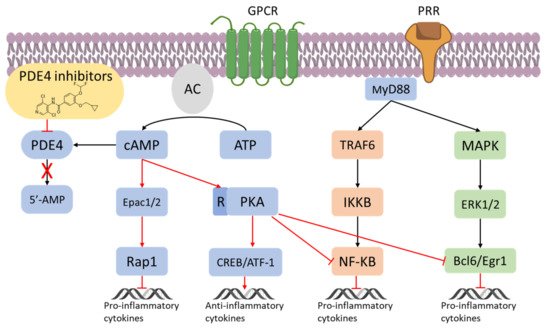
The Nuclear Factor κ-light-chain-enhancer of activated B cells (NF-κB) can mediate cell-specific responses and pharmacological attempt to block its activation is being considered a new therapeutic option in inflammatory conditions (
Figure 3) [
36,
37,
38]. It is known that c-AMP interferes with the NF-κB signalling, being in parallel recognized as an immunosuppressive and anti-inflammatory actor as well [
39]. As a result, PDE4 could be potentially useful to indirectly leverage the inactivation/activation of the NF-κB signalling in inflammation. For instance, in endothelial cells, the high levels of c-AMP produced by an adenylate cyclase activator (e.g., forskolin) have been reported to prevent the NF-κB-dependent gene transcription [
40]. When macrophages are treated with ethanol chronically, the inhibition of PDE4 has been proved to decrease the TNF-α mRNA expression, by the intervention of the transcriptional modulation of NF-κB [
41]. Moreover, the inhibition of PDE4 leads not only to less NF-κB-mediated TNF-α expression but also to activation of the PKA and increased synthesis of IL-10 [
42]. Consequently, inhibitors of PDE4 may be useful to modulate, negatively or positively, gene expression. Variation in the NF-κB by PDE4 and c-AMP has also been reported in T cells, where PDE4 has been found to control the proliferation of T lymphocytes, along with the concentration of TNF-α and other interleukins such as IL-2, IL-4, and IL-5 [
43]. The c-AMP has also been reported as a crucial mediator of the regulation of T cell suppression, by crossing the cell membrane of responder T cells, such as CD4-positive [
44] and TH2 subsets [
45], as well as inhibiting T cell proliferation. In the context of inflammation, PDE4 are known to promote also chemotaxis and degranulation in both eosinophils and neutrophils. These effects are mediated by the increased concentration of IL-8, leukotriene B4 and superoxide anion stimulated by PDE4 in neutrophils. Moreover, PDE4 has been reported to control the expression of adhesion molecules, such as the β2-integrin Mac-1 in neutrophils, resulting in augmented adhesion to vascular endothelial cells [
46,
47].
Figure 3. PDE4 inhibitors in the regulation of inflammatory responses [
36,
37,
38].
4. From In Vitro and Preclinical Profile of The First PDE 4 Inhibitors to Approved Drugs
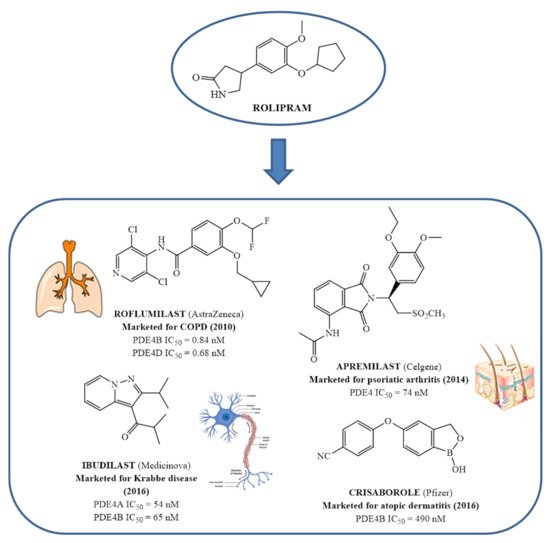
In the context of PDE4 inhibitors, it would appear definitely appropriate to start with Rolipram (
Figure 1), the synthesis of which dates back to 1977 [
48,
49]. Rolipram has certainly been the most investigated PDE4 inhibitor, from the first studies carried out shortly after its synthesis, to investigations conducted at present time [
50,
51,
52,
53,
54,
55,
56], including further clinical trials (as detailed below). Already at the time of the first studies on Rolipram and its analogues, there was a body of experimental evidence (confirmed later on) that PDE4 inhibitors were able to suppress inflammatory and immunomodulatory responses in a variety of murine and human cells, to block superoxide generation in monocytes, macrophages, neutrophils and eosinophils, to reduce TNF-α release in monocytes and macrophages, and to suppress chemotaxis and phagocytosis [
57,
58]. It is important to highlight that in eosinophils, which are the effectors “par excellence” of asthma, PDE4 inhibitors can suppress superoxide generation, chemotaxis, degranulation, LTC4 synthesis and CD11b expression. In vivo, PDE4 inhibitors demonstrated bronchodilatory effects and the ability to reverse bronchospasm induced by a variety of agents, thus, the profile of selective PDE4 inhibitors appeared to fulfil the requirement for the treatment of inflammation-based pathologies such as asthma and chronic obstructive pulmonary disease (COPD) and some auto-immune diseases [
15,
59,
60,
61,
62].
Even though such promising in vitro and preclinical results generated a large consensus on the concept of PDE4 as a valid target for the treatment of the above diseases, clinical evaluation of several potent and selective PDE4 inhibitors was strongly disappointing [
63]. A lack of correlation between preclinical and clinical data was a prominent limitation. In this context, it is worth mentioning that the first clinical studies were performed with the first generation of PDE4 inhibitors (rolipram and congeners), which were characterized by serious adverse reactions found, at least in part, associated with the affinity for the high-affinity Rolipram binding site (HARBS) [
64]. Only in 1996, it become evident that a better side-effect profile could be obtained with agents preferentially targeting the catalytic site over HARBS [
65]. Then, this type of selectivity was not pursued any longer, and another type of selectivity versus the gene products A-D was proposed after these subtypes were characterized, their tissue distribution demonstrated and their different functional role suggested [
66]. In the early 2000s, the PDE4D isoform was indicated as being responsible for PDE4 inhibitor-induced emesis [
67], although recent studies suggest that it may not be the only factor involved in this side effect [
68]; on the other hand, it has been demonstrated that PDE4B selective inhibitors produce potent anti-inflammatory and reduced emetic effects [
69].
In the same way, crucial advancements were made on the knowledge of the biochemistry, pharmacology and molecular biology of the PDE4 family, including a detailed characterization of the different PDE4 subtypes, differentially expressed in tissues and cells [
66]. The identification of a variety of potent and selective compounds, defined as second-generation PDE4 inhibitors, sustained the hope that the lower emetic potential of these molecules should overcome the problems encountered with rolipram and its congeners, whose development as antiasthma drugs failed due to the adverse reactions [
16]. This newly acquired information greatly stimulated the research leading to the synthesis of Roflumilast by AstraZeneca, which was finally approved as a COPD drug in the EU (in 2010) and in the USA (in 2011) [
70]. Since the marketing of Roflumilast, three additional compounds have been marketed as PDE4-inhibitor drugs (
Figure 4) i.e., Crisaborole (by Pfizer) for atopic dermatitis [
71], Apremilast (by Celgene) for psoriatic arthritis [
72] and Ibudilast (by MediciNova) for Krabbe diseases (also known as globoid cell leukodystrophy) [
73]. However, research in the field of PDE4 inhibitors has remained very active, leading to very potent and pharmacologically relevant ligands that have entered clinical trials for the treatment of a range of diseases. This review aims to provide an overview of the most interesting compounds that have been developed and entered into clinical trials since the marketing of the first PDE4 inhibitor Roflumilast.
Figure 4. From Rolipram to marketed PDE4 inhibitors.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27154964