1. History and Evolution
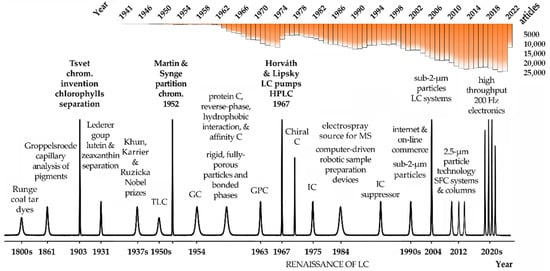
German chemist Friedlieb Ferdinand Runge, the scientist that discovered caffeine and quinine, was also the first to use chromatography principles in the early 1800s (
Figure 1). Runge was interested in the production of dyes and bleaches from coal tars, and he used filter paper to obtain color separations from those mixtures [
2,
3]. The color patterns that Runge obtained (self-grown pictures) are considered an incipient form of paper chromatography, despite Runge not describing any theoretical considerations to support his findings. In 1861, Groppelsroede used the capillary properties of water to separate colored pigments. To perform this capillary analysis, he dipped one end of the paper strip into an aqueous solution [
3,
4]. Groppelsroede, similarly to Runge, did not report a satisfactory explanation for such separation resembling paper chromatography. In the early 1900s, however, Mikhail Tsvet used a hydrocarbon solvent and a carbohydrate powder as a stationary phase to separate chlorophylls and plant pigments from green leaves. The colored bands Tsvet obtained certainly led him to name the technique he invented chromatography, in that case, adsorption chromatography. Curiously, his surname, Tsvet, means color in Russian. Tsvet has made hundreds of experiments and he was the first scientist to explain the fundamentals of chromatography [
5] (
Figure 1). Most of the principles Tsvet described are still applicable nowadays in modern chromatography. For that reason, he is known as the father of chromatography. Unfortunately, the seminal work of Tsvet was only published in Russian, and that prevented its fast dissemination. For that reason, the next hallmark in chromatography development did not occur until almost three decades later. In 1931, Lederer and collaborators achieved the separation of lutein and zeaxanthin in carbon disulphide and the xanthophylls from egg yolk on a column of calcium carbonate powder 7 cm in diameter [
6]. Several years later, flow-through chromatography became an important laboratory separation technique. The work of Khun, Karrier, and Ruzicka, awarded with the Nobel Prize in 1937, 1938, and 1939, respectively, for their work on chromatography, was another major hallmark of chromatography evolution. The next important milestone, eventually one of the most important, was made by biochemists Archer Martin and Richard Laurence Millington Synge, awarded the Nobel Prize in Chemistry in 1952 for the invention of partition chromatography. The original process developed by Martin and Synge to isolate acylated amino acids from protein hydrolysates by extraction of the aqueous phase with a chloroform organic phase was very tedious, but it boosted future and unprecedented improvements in chromatographic resolution. Soon, Martin’s and Synge’s proposal was improved with a chromatographic column containing silica gel particles and later cellulose [
7] (
Figure 1). Since then, chromatography developed rapidly as well as in several directions beyond liquid–liquid and gas–liquid partition chromatography. During the 50s, for instance, the paper was substituted by thin layers of silica gel, originating the thin-layer chromatography (TLC), and the initial simple and well-defined mixtures containing small molecules gave rise to more complex biological systems. At another level, protein chromatography underwent a significant evolution using technologies such as reverse-phase, hydrophobic interaction, and affinity chromatography.
Figure 1. Overview of major milestones in the evolution of chromatography. The number of articles published in the literature according to PubMed servers and using the keyword chromatography are indicated in the top part of the figure. Legend: C—chromatography, GC—gas chromatography, GPC—gel permeation chromatography, HPLC—high-performance liquid chromatography, IC—ionic chromatography, LC—liquid chromatography, MS—mass spectrometry, SFC—supercritical fluid chromatography, TLC—thin-layer chromatography.
In its initial development, column liquid chromatography was hampered by the very slow separation process caused by mobile phases crossing 100 µm sorbent particles in the column only by gravity. To achieve faster separations, Csaba Horváth and Seymour Lipsky of Yale University introduced the use of pumps. In 1966, they published a paper describing an ion-exchange separation of organic compounds [
8], and one year later they introduced fast liquid chromatography (LC) using a pump operating at higher pressures than in their previous work [
9]. Horváth is generally credited with building the first high-pressure liquid chromatography instrument [
10]. Furthermore, effectively during the decade 1970, the introduction of pumps allowed a major increase in the mobile phases flow and the definition of high-pressure liquid chromatography [
5]. Meanwhile, durable adsorbents with much lower particle sizes between 5 and 10 μm, high-pressure pumps up to 400 atm, and flow-through detection systems were developed, and high-pressure was substituted by high-performance liquid chromatography (HPLC), which is how the HPLC acronymous is still currently used [
5]. Soon, HPLC surpasses gas chromatography (GC) in terms of applications and importance. This period was known as the renaissance of LC [
5]. In the late 1970s, GC took advantage of the development of fused silica capillaries, which dramatically improved its performance and efficiency.
However, GC was limited to the analysis of volatile compounds and many important analytes could not be derivatized and made volatile. Therefore, by the 1980s, HPLC was already used in most laboratories worldwide and progressive improvements in sensitivity, speed, accessibility, and resolution continue to be made. The capacity to separate very similar compounds, including chiral compounds, should also be mentioned as an important achievement of chromatography as an analytical tool. Chiral chromatography was boosted by the work of Davankov with chiral ligand exchange chromatography, a method for separating optical racemate isomers [
11].
The introduction of new detectors, first using UV/IR spectrophotometry, and then mass spectrometry (MS), was another hallmark in chromatography evolution. The use of MS detectors allowed a major improvement in the detection limits and identification of compounds in many fields of research, particularly in protein analysis.
Ion chromatography (IC) also met considerable improvements, the most notable the use of the suppressor. This strategy removes the eluent, therefore, limiting the background and reducing noise, and favors the conversion of the analytes into more conductive forms, resulting in a major gain in sensitivity of the IC.
Additionally, very relevant are the developments of particle technology and column chemistries, from the initial big and fully porous particles to the superficially porous materials, currently under 2 µm in diameter. This was essential to introduce new column features, lower dispersibility, and enhanced binding capacity to respond to the high throughput, speed, and resolution requirements of the biopharmaceutical industry. The successive developments in chromatography requested faster and more powerful processing units and connections and this would be not possible with the development of faster computers and internet connections to process and share the growing amount of data. The first computers and software to work with chromatography were only introduced in the late 1980s and they were a remarkable evolution. Modern electronic devices and chromatographic configurations can produce, process, and transfer data at collection rates of up to 200 Hz. This means thousands of samples being processed each day, something that was unimaginable only a few years ago. Figure 1 condenses some of the most important features and hallmarks in chromatography since its invention.
Currently, HPLC is almost omnipresent in any field of research and industry and continues to evolve toward miniaturized, faster, and more efficient separations, using more comprehensive and sensitive analytic sensors, and user-friendly analytical platforms to push the limits of high-resolution and high-throughput even further. Capillary GC or simply GC is also still frequently used, but only for volatile and semivolatile analytes, while the other forms of chromatography, despite some advances, such as the high-performance TLC (HPTLC), are used to a lesser extent.
Chromatography has been essential in establishing many important therapies for patient care and, certainly, this will be a field where it will continue to evolve. New treatments involving single cell and gene therapies are at the forefront of healthcare and chromatography will be fundamental to bringing new drugs and treatments to the market faster than any other analytical procedure can deliver [
10].
2. Principle of Chromatography and Types of Chromatography
As an analytical technique, chromatography has contributed to facing and solving various societal challenges, being used to separate, isolate, and purify the components of a mixture for qualitative and quantitative purposes. Chromatography is a biophysical-chemical approach of separation where the target analytes in a mixture are smeared onto a solid in the form of a porous bed, bulk liquid layers, or films (stationary phase) and separated with the help of a mobile phase (e.g., liquid, gaseous) that percolates through the stationary phase. During the migration of the fluid through the stationary phase, different transport phenomena, which include diffusion and flow anisotropy, will occur. The relative interaction of a solute with these two phases is expressed by the partition (K) or distribution (D) coefficient, which corresponds to the ratio between the concentration of solute in the stationary phase and the concentration of solute in the mobile phase. These differences result in the retention of some components in the stationary phase that will move slowly in the chromatography system [
12].
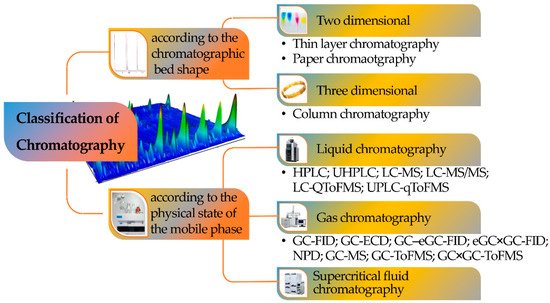
Chromatography can be classified according to the mobile phase, the shape of the chromatographic bed, or the stationary phase, with the mobile phase being the most important criterion since it defines the class of samples that can be investigated with each one of the chromatographic versions (Figure 2).
Figure 2. Classification of chromatography according to the chromatographic bed shape and the physical state of the mobile phase.
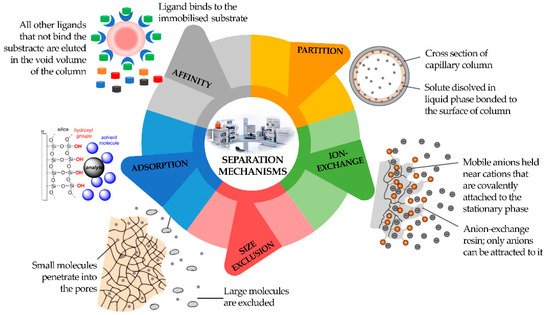
Consequently, a division can be made among LC, supercritical fluid (SFC), and GC. Chromatography can also be defined according to four different sorption mechanisms, namely surface adsorption, partition, ion exchange, and size exclusion (
Figure 3). In the adsorption process, the stationary phase is a polar adsorbent, normally silica (slightly acidic) but also alumina (slightly basic), charcoal (nonpolar), or several other materials, while the mobile phase is nonpolar (usually a solvent with polarity within the range from hexane to esters). The mobile phase competes with the sample analytes for adsorption at the active sites of the stationary phase. Due to weak interactions with the stationary phase, nonpolar analytes are eluted first, followed by analytes of increasing polarity. It is the method of choice for isomer separation due to the steric properties of the sample compounds. The intermolecular forces primarily responsible for chromatographic adsorption include van der Waals forces, electrostatic forces, hydrogen bonds, and hydrophobic interactions [
13].
Figure 3. Classification of different types of chromatography according to separation mechanism.
The partition chromatography mechanism is based on a thin film formed on the surface of a solid support by a liquid stationary phase. Solute equilibrates between the mobile phase and the stationary liquid. In ion exchange chromatography, the ion exchanger groups can be bonded to silica or polystyrene. It is based on ionic equilibria between solute, buffer, and stationary phase ions and counter-ions.
In size exclusion chromatography, the sample analytes will be separated according to their size. They will not be retained by the column packing but will enter the pores where the mobile phase is stagnant. Large molecules can use a smaller fraction of the pores’ volume than small molecules and will be eluted earlier. Analytes that are bigger than the pores are excluded and will appear as the first fraction at the column end [
12].
Nevertheless, a suitable sample preparation procedure prior to chromatography analysis is crucial, since direct analysis can result in low sensitivity, accuracy, and reproducibility due to the existence of interferents in the sample matrix. In this sense, to achieve high-quality analytical data with high accuracy, reproducibility, selectivity, and low sensitivity limits, the sample preparation procedure should include steps such as fractionation, isolation, and enrichment of the target analyte. Such an endeavor has been achieved through the improvement of the properties of existent materials, as well as the discovery of new ones, such as ionic liquids, graphene-derived materials, molecularly imprinted polymers, magnetic nanoparticles, carbonaceous nanomaterials, among others.
2.1. Gas Chromatography
GC is a popular analytical approach used to separate and analyze samples that can be vaporized (vapor pressure at temperatures below 350–400 °C) without thermal decomposition. In GC, the components of a sample are dissolved in a solvent and vaporized to separate the analytes by distributing the sample between stationary and mobile phases. The mobile phase is a chemically inert gas (e.g., helium, hydrogen, nitrogen) that transports the target analytes through the heated column. A constant flow rate of carrier gas is crucial since it significantly influences the separation efficiency and the quantification of results [
12].
Packed and capillary (also known as open tubular) columns with different dimensions are the most used columns for GC. Packed columns comprise finely divided, inert, solid supports (e.g., diatomaceous earth, fluorocarbons, graphitized carbon black, and glass beads) coated with the liquid stationary phase. Packed columns present advantages compared to capillary columns since they have between 10 and 1000 times better sample capacity, which requires a large amount of sample. Nevertheless, packed columns have from 25–50% fewer theoretical plates per meter (m) than capillary columns. A capillary column is a glass or fused-silica tube of very small internal diameter (usually between 0.20 and 0.53 mm). The inner surface of a capillary column is coated with a thin layer of stationary phase, so it is still possible for the solute molecules to contact the inner walls of the tubing. The high separation efficiency is one of the advantages of a capillary column, allowing peak resolution. In addition, the separation efficiency can be increased through the application of a temperature gradient instead of an isothermal separation mode. After the elution of the column, the target analytes can be detected by several detectors, such as mass spectrometry (MS), flame ionization detector (FID), electron capture detector (ECD), nitrogen and phosphorous detector (NPD), among others [
14,
15]. FID detectors are usually employed in portable GCs. These configurations are used to separate and analyze target analytes which can be vaporized without decomposition. Overall, FID detectors present several advantages such as reliability, versality, ease of operation, and simplicity. In addition, FID shows small or no signal for common carrier gas (e.g., He, Ar, N
2) or common contaminants (e.g., O
2, H
2O). The main drawback of these detectors is its destructive nature and inability to provide structural information of target analytes. The MS detector is another detector often coupled to GC, allowing to elucidate the molecular mass and molecular structure of identified and/or quantified compounds. MS can be used without coupling to GC, but the interpretation is more difficult, mainly when complex mixtures are analyzed. To overcome this problem, MS can be used in tandem with other MS detectors.
2.2. Liquid Chromatography
The mobile phase of LC is a liquid (organic solvent, water, and/or a solution of two or more organic solvents and water) and there are several configurations regarding the instrumental as well as the separation steps, such as paper, TLC, and HPLC.
Paper chromatography is the simplest and most cost-effective LC technique, in which the chromatographic bed comprises a paper (e.g., cellulose) shoaled in a liquid solvent that acts as a mobile phase. The samples are applied a few centimeters from the end of the paper and impregnated with some suitable solvent. The solvents penetrate the paper through capillary action (depending on whether ascending or descending development is applied), and, in passing over the sample spot, transport along with it the components of the sample. Those components move with the flowing solvent at velocities that are dependent on their solubilities in the stationary and flowing solvents. In the end, the paper is removed from the developing chamber and the separated zones are detected by applying a suitable method [
12].
The TLC is more versatile than the paper chromatography used to separate non-volatile compounds, since several different stationary phases are available, such as cellulose, silica, and alumina. The separation is based on the affinity between the target analytes and the adsorbent that will appear as individual spots on the paper after the chromatographic separation. TLC is an analytical approach broadly used since it is simple, cost-effective, fast, and has good reproducibility and high sensitivity [
16]. In addition, high-performance TLC, designed as HPTLC, that uses 5 and 10 μm stationary phase particles, allow for higher separations.
HPLC is an up-to-date application of LC. HPLC pumps the target analyte dissolved in a solvent (mobile phase) at high pressure through a column with an immobilized chromatographic packing material (stationary phase). The retention time of the target analyte depends on the sample and solvent properties, as well as the stationary phase. Thus, the analytes that have the highest interaction with the stationary phase exit the column at the end and, consequently, they display higher retention times.
Diverse solvent combinations can be used as mobile phases and can also contain water and/or organic components. The HPLC column is frequently a stainless-steel tube ranging from 50 to 250 mm in length and from 1–4.6 mm in diameter, packed with chemically modified silica particles (<1–5 μm in diameter). The microparticle packing (low diameter compared to conventional HPLC columns) contributes to the improved resolution of the mixture, requires low consumption of the mobile phases and samples, and can easily be used in tandem with MS due to the lower flow rates involved. There are several stationary phases available for HPLC separations, with silica particles modified with C18 groups the most used. In addition, the diversity of column diameters, length, particle size, and solvent selection contributed to the huge number of choices commercially available to the researcher [
17].
HPLC analyses can be carried out in a diversity of modes: (i) reverse phase chromatography (the most important), in which the stationary phase is nonpolar, and in most cases hydrophobic (C18-modified silica); (ii) normal phase chromatography, where the stationary phase is hydrophilic (silica); (iii) hydrophilic inter-action chromatography; (iv) ion chromatography, where the stationary phase is an ion exchange material for cationic or anionic analytes. The mechanisms responsible for distribution between phases include surface absorption, ion exchange, relative solubilities, and steric effects. After the elution from the column, the target analytes can be detected by several detectors, such as MS, photodiode array (PDA), fluorescence (FLD), refractive-index detector, among others. The main disadvantages of PDA and FLD are the restricted number of target analytes with absorbance or fluorescence properties, and the limited sensitivity which the low injection volumes applied allow to obtain. To overcome this problem, LC is often used tandem with MS. This detector has the advantage of unequivocal mass identification and provides structural information of the target analytes. This coupling was made possible due to the development of atmospheric pressure ionization (API) sources, such as electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). HPLC has the advantages of high sensitivity, accuracy, separation efficiency, and wide application range, particularly for the detection of high boiling point and non-volatile compounds [
12].
3. Major Achievements
One of the most crucial changes in chromatography is the reduction of time analysis without losing the resolution. In this sense, fast-GC appears to have improved their predecessors, namely concerning rapid oven heating and cooling, extended inlet pressures, support for hydrogen carrier gas, and faster detector response time. This was possible because several mechanisms were adopted in fast-GC, such as adjusting column dimensions, higher flow of carrier gas, lowering the pressure at the chromatographic outlet and increasing the heating temperature in the programmed temperature [
18]. Perez-Palacios et al. [
19] established the optimum combination of parameters (methanol + chlorotrimethylsilane, lyophilized samples, and oven heating) to achieve the quantification of the highest possible amount of fatty acids in meats, reducing the time of a GC run from 60 to 10 min. Paolini et al. [
20] developed a fast GC–FID method for the analysis and quantification of 16 volatile compounds belonging to different chemical classes in alcoholic beverages, using a chromatographic run of only 8 min. Fialkov et al. [
21] achieved reasonably good separations with full analysis cycle times of less than 1 min by combining, for the first time, low-pressure (LP) GC–MS with low-thermal mass (LTM) resistive-heating for rapid temperature ramping and cooling of the capillary column. This column configuration in LTM-LPGC-MS trades a 64-fold gain in speed of analysis versus standard GC–MS and a 4-fold loss in chromatographic peak capacity, thereby converting analysis time from minutes into seconds in common applications.
Regarding LC, the reduction in the internal diameter of the column as well as of the particle size of the stationary phase contributed to improving the separation of several target analytes. Ultra-high performance liquid chromatography (UHPLC) appeared as a greener approach to LC, since it requires a lower solvent consumption and offers greater chromatographic resolution and higher sensitivity while requiring a shorter analysis time [
22]. Zhao et al. [
23] compare the efficiency of HPLC–MS/MS and UHPLC–MS/MS analytical platforms in the simultaneous determination of eight coccidiostats in beef. The data obtained showed that UHPLC–MS/MS was more efficient, faster, and consumed less mobile phase than HPLC–MS/MS. In addition, despite the eight target analytes could be completely separated from impurities using the two methods, the sensitivity of the UHPLC–MS/MS was higher than that of HPLC–MS/MS. The Brazilian green propolis phenolic profile was established using UHPLC-ESI-QToFMS and HPLC–MS. The data obtained showed, for the first time, three different isomers of isochlorogenic acid identified using UHPLC-ESI-QToFMS, indicating the precision and accuracy of this analytical approach [
24]. More recently, miniaturized LC versions, such as microflow LC (internal diameter 0.5–1 mm), capillary liquid chromatography (100–500 μm), and nano liquid chromatography (NanoLC, <100 μm), improved sensitivities in comparison to LC [
18]. Ponce-Rodríguez et al. [
25] assessed the performance and operability of a portable NanoLC in the determination of several methylxanthines in water samples. In addition, a comparative study was also performed between in-tube solid-phase microextraction (IT-SPME) coupled with capillary liquid chromatography (CapLC) or NanoLC. The data obtained showed that IT-SPME/portable NanoLC-based methods were much better in terms of chromatographic resolution and organic solvent consumption per sample, using only 200 μL versus 10 mL for IT-SPME-CapLC. Piendl et al. Of note, Piendl et al. [
26] introduced the online hyphenation of chip-based high-performance liquid chromatography (chipHPLC) with ion mobility spectrometry (IMS) via fully integrated electrospray emitters. The method showed promising results, highlighting the potential of IMS as a detection technique for chip configurations, adding the advantages of simplicity, portability, economy, and robustness. Lam et al. [
27] proposed a hand-held, battery-powered, portable LC system with a high sensitivity UV-LED-based z-cell detection (HSDC) and compared it with on-column detection strategies used in previous studies. The HSDC flow-cell-based detector showed excellent sensitivity, with low stray light levels, and negligible heat effects using low input currents (<20 mA).
Another challenging trend to improve the separation power is multidimensional chromatography, with GC × GC and LC × LC the most popular. Nevertheless, one of the main drawbacks of this improvement compared to other chromatography approaches (e.g., fast-GC) is the time of analysis required [
13]. Multidimensional chromatography is an analytical approach which can deliver heightened separation performance for complex and difficult substances. This is obtained by passing the sample through two different separation stages, applying multiple columns, each with a different stationary phase. Whiting et al. [
28] described the development and evaluation of micro-comprehensive two-dimensional gas chromatography (GC × GC) constituted by microfabricated columns and a nanoelectromechanical system resonator as a detector. The resulting system allows eco-friendly and ultrafast chromatographic separation. Acevedo et al. [
29] described two-dimensional separation (LC × LC) pioneering applying sequential injection chromatography (SIC) to enhance the performance of the analysis of aromatic biogenic amines. The proposed analytical approach led to a cost-effective, greener, and faster chromatographic separation without involving additional mobile phases and energy consumption. However, in multi-dimensional LC, the robustness of the method is not very good, since the eluting portion of the first column was not fully compatible with the second column, resulting in peak dispersion and retention time shifts.
Finally, it is interesting to note that the basic principles that determines the separation of analytes in the simplest chromatography modes, such as paper and TLC, are the same principles that determine the separation in high-resolution and high-performance chromatography approaches, such as GC × GC-ToFMS or the UHPLC-QToFMS. However, the technological evolution from the former to the recent techniques is immense, namely in terms of the nature and particle size (submicron) of the stationary phases, enabling highly efficient separations, with high-resolution power and short analysis time. Such configurations also involve efficient detection systems with extremely low sensitivities that allow the detection of trace levels as low as femtomole. In addition, the evolution of chromatography data systems (CDSs) in the last decades also potentiated even further the contribution of chromatography to the progress of several fields of science. In fact, the significant evolution of CDSs in the last decades is remarkable. In the 1970s, we were in the “stone age”, where the chromatographic signals were recorded on chart paper followed by peak quantification using manual peak measurement, by “cutting and weighing” the actual peaks. In contrast, in the current “technologic age”, the automation of peak measurement and instrument control, and CDSs data upload and storage in readily shareable online databases is a reality.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27165267