Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Metal–organic frameworks (MOFs)—typically formed from metal ions/clusters bridged by multidentate ligands in an extended framework—have provided solutions to tackle challenges in areas such as catalysis. By either integrating porphyrins/metalloporphyrins inside pores freely in situ or by grafting on the surface using post-synthetic methods and/or as part of the network component, porphyrin-based MOFs could be easily constructed.
- metalloporphyrins
- metal–organic frameworks
- porphyrins
- catalyst
- synthesis procedures
1. Introduction
The recently emerged porous materials, metal–organic frameworks (MOFs)—typically formed from metal ions/clusters bridged by multidentate ligands in an extended framework—have provided solutions to tackle challenges in areas such as catalysis [1][2][3], gas storge/separation [4][5], biomimetic applications [6][7][8], drug delivery [9][10][11], electrochemical applications [12][13] and biomedical chemistry [14][15]. Moreover, the structures can be tuned by replacing or incorporating specific linkers or suitable unsaturated metal ions, in addition to adjusting the pore size and/or geometry which substantially influence their catalytic behaviors toward many substances participating in the reaction [16][17]. Amongst the main categories of MOFs, porphyrin-based MOFs have demonstrated themselves as a tangible material able to provide the properties of both MOFs and metalloporphyrin complexes in one scaffold [18]. Despite being synthesized less frequently than other types of MOFs and less often explored in research, they have had considerable impact on multiple fields, particularly biomimetic and biomedical ones as catalysts, owing to their resemblance to some molecules discovered in nature [19].
Tetrapyrrole ligands such as porphyrin (or porphine in the unsubstituted form) and related macrocycles chlorin and corrin are naturally occurring in several bioinorganic metal complexes. The ability of the planar or nearly planar tetradentate ring system to stabilize kinetically labile metal centers (i.e., MgII, NiII, FeII/III, CoII) results in the selective formation of stable metal complexes containing an extensively conjugated π system (Figure 1).
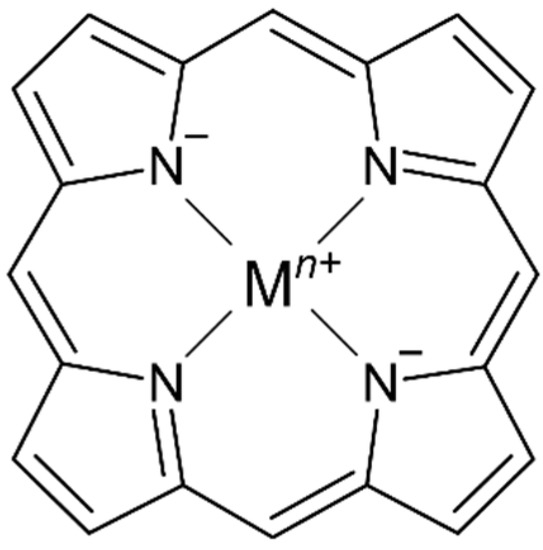
Figure 1. Representative structure of a metalloporphyrin complex with a porphine ligand core.
Thus, metalloporphyrins, due to their abundance in nature, have been explored during the last decades. The synthetic bioinspired complexes resemble in structure, central atoms, and properties the most common naturally occurring biomolecules such as hemoglobin which transports oxygen in animal bodies, chlorophyll which acts as a light-scavenging antenna in photosynthesis inside plants, or vitamin B12 which is important for metabolism in the cells (Figure 2, Table 1) [20]. The combination of such favorable properties makes artificial metalloporphyrins highly suited for applications in photosynthesis [21], electrochemical [22], biosensing [23], biomedical [24] applications for tumor therapy [25] and bioimaging [26].
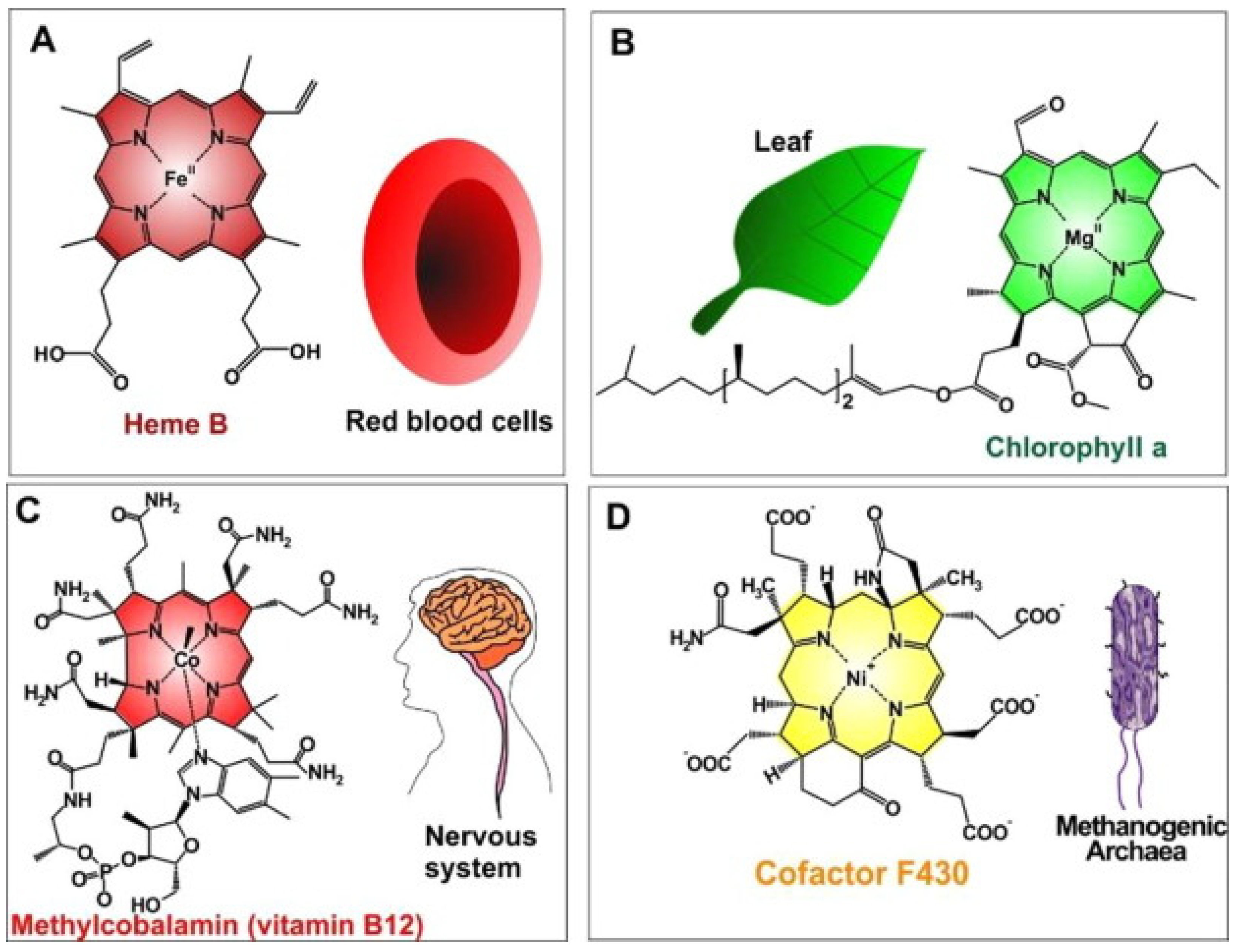
Figure 2. Naturally occurred MPs (metalloporphyrins) (A) iron(II)-porphyrin “Heme B in RBCs” to convey oxygen; (B) magnesium(II)-porphyrin “chlorophyll a” needed for plant photosynthesis; (C) cobalt(II)-porphyrin “methylcobalamin (as vitamin B12)” assisted to facilitate nerve system performances; (D) nickel(II)-porphyrin “Cofactor F430” accelerates methanogenesis in methanogenic archaea). Reprinted with permission from [20].
Table 1. Naturally occurring metalloporphyrin complexes [27].
| Metal Ion | Ionic Radius (ppm) * | Naturally Occurring Complex |
|---|---|---|
| Mg2+ | 72 | Chlorophyll |
| Ga3+ | 62 | Gallium(III) porphyrin complexes have been found in crude mineral oil but not in living organisms |
| (V=O)2+ | ≈60 | Vanadyl porphyrins are relatively abundant in certain crude oil fractions but they have not been observed in living organisms |
| Fe2+ high spin Fe2+ low spin Fe3+ high spin Fe3+ low spin |
78 (too large) 61 65 55 (rather small) |
Fen+ in various oxidation and spin systems is present in heme systems such as hemoglobin |
| Co2+ | 65 | Cobalamins (vitamin B12) |
| Ni2+ | 73 | Cofactor F430 (catalyzes the reaction that releases methane in the final step of methanogenesis in archaea), tunichlorin |
* The ideal ionic radius for a proper in-plane coordination is 60–70 ppm. There have also been prepared many artificial complexes containing mostly Mn3+ (≈60 ppm), Cu2+ (73 ppm) and Zn2+ (74 ppm).
The crystal engineering of MOFs based on metalloporphyrins, or tetrapyrrole ligands in general, began in the early nineties [28]. The intercalation of tetrapyrrole ligands was recently recognized to be driven mostly by weak dispersive forces and either an offset or a proper π−π stacking with other components of the MOF [29][30][31][32][33]. Interestingly, there is usually a lack of a stronger specific interaction between the porphyrin sheets themselves. Therefore, the construction of a crystalline MOF depends very much on the additives and their ability to provide suitable interactions and binding with metalloporphyrin and/or other components [34][35][36][37][38].
On the other hand, further to metallization in their center [39], metalloporphyrin complexes can also be additionally peripherally functionalized at meso- or β-positions [40] (in Porphyrin, there are typically 12 positions that can be exchanged in the environs (Pyrrolic rings containing the eight β positions and the other four meso ones which are attributed to the methine substituents)) or even complexed by various (non-) transition metals providing versatile moieties which had led to their use as spacers to construct several porphyrin-based MOFs where they could serve either inside the pores (porphyrin@MOFs) [41] or as linker merged in the architecture throughout the framework (porphyrinic MOFs) [42]. The representative structures of synthetized derivatives of porphine are depicted in Figure 3. They benefit from the extended π conjugated system throughout the planar molecule, which moderates substitution of the metal ions and the functionalities of porphyrin itself [43]. Therefore, it illustrates exceptional electrochemical and photophysical exploitations while possessing an extraordinary chemical and physical durability. Additionally, porphyrins and their accessories normally have the strongest Soret band (400–450 nm) and a pack of steadily reduced Q-bands located somewhere in the range of 500 to 700 nm in the absorption spectrum [44]. These features have led them to be considered as one of the most significant organic chromophores with remarkable adsorption bands in the visible region.
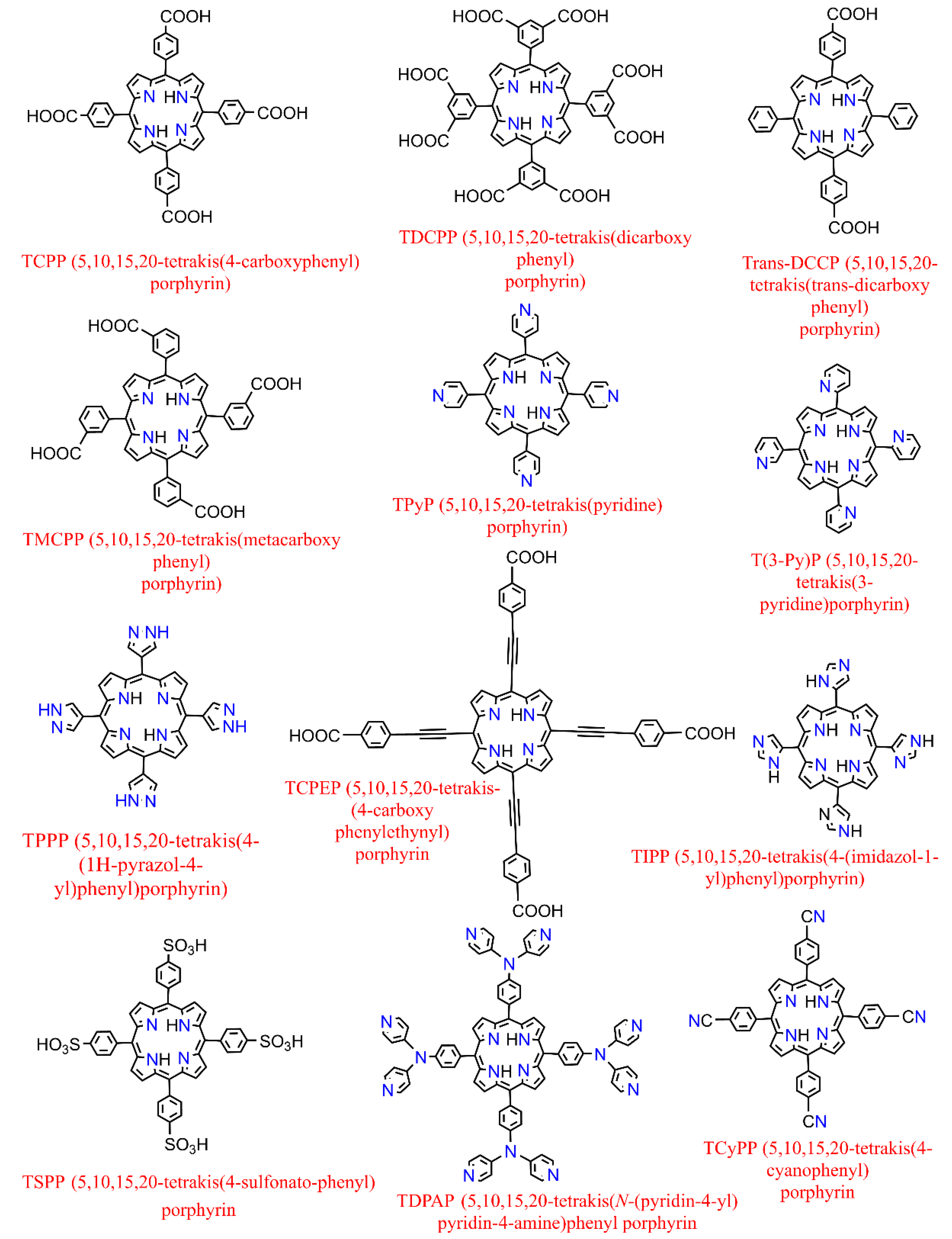
Figure 3. Examples of some of the previously fabricated porphyrin linkers.
2. Synthesis Procedures of Porphyrin-Based MOFs
By either integrating porphyrins/metalloporphyrins inside pores freely in situ or by grafting on the surface using post-synthetic methods and/or as part of the network component, porphyrin-based MOFs could be easily constructed. However, downsizing MOFs to the nanoscale will even more profoundly develop their size-dependent properties when encapsulating or accompanying such proactive molecules for any related applications [14].
2.1. In Situ Method of Porph@MOFs Synthesis
Inspired by these facts, some promising routes to combine porphyrin derivatives into MOF frameworks emerged such as porphyrin@MOFs (porph@MOFs). These include the entrapment of (metallo-) porphyrins into the cavities or the decoration of the surface of MOFs where the former method can be performed using one-pot fabrication in situ, and the latter can be executed post-synthesis. In contrast to in situ assembly in which free porphyrin basis/metalloporphyrin are entrapped by MOF precursors (metal ion and ligand) by self-assembly simultaneously (ship-in-a-bottle) [45], the post-synthetic method which occurs by anchoring to the exterior or the inclusion of them inside the MOF pores is mainly based on weak chemical interactions such as hydrogen bonding, electrostatics, van der Waals forces and others, between the pre-obtained MOF and porphyrin base/metalloporphyrin [46]. The simplicity and straightforwardness of porphyrin entrapping by in situ formation led to this path being applied extensively by many researchers working in this field even though the post-functionalization requires the acknowledgement of some issues such as the creation of a suitable interaction between these two materials [14][20]. Parameters that should be considered before postsynthetic fabrication include the activation of MOF pores and channels by guest solvent removal inside the structure during synthesis to allow for the incorporation of porphyrin instead, the dimensions in terms of shape and size of the porphyrin encapsulated into the cavities should be appropriate, and the stimulus required to initiate bond construction between the porphyrin and MOF structure.
As described above, embedded porph@MOF can be prepared by the in situ mixing of pre-synthesized porphyrins and MOF reactants (metal ion salt and linkers). A series of metalloporphyrin-decorated Cu-based MOFs with a coral-like shape (named as M-TCPP@Cu) were obtained using a one-pot reaction strategy [47]. Accordingly, the resulting MOFs were developed through intermediate enrichment-enhanced conversion to assist the electrochemical reduction of CO2 to C2H4. The respective porph@MOFs were obtained by dissolving H3BTC and TCPP/M-TCPP in a mixture of ethanol and DMF followed by the addition of Cu(NO3)2·3H2O in aqueous solution in situ to produce M-TCPP@Cu-MOF (M = Fe, Co, Ni). An ionic Mn-metalloporphyrin was reported which is presented in Figure 4 that was encapsulated into the interior pores of ZIF-8 by a simple method through which all the precursors in DMF were sealed in a Teflon-lined autoclave and heated at 140 °C for 2 days [48]. The crystals were then used as heterogeneous catalysts for a cycloaddition reaction of CO2 with epoxides.
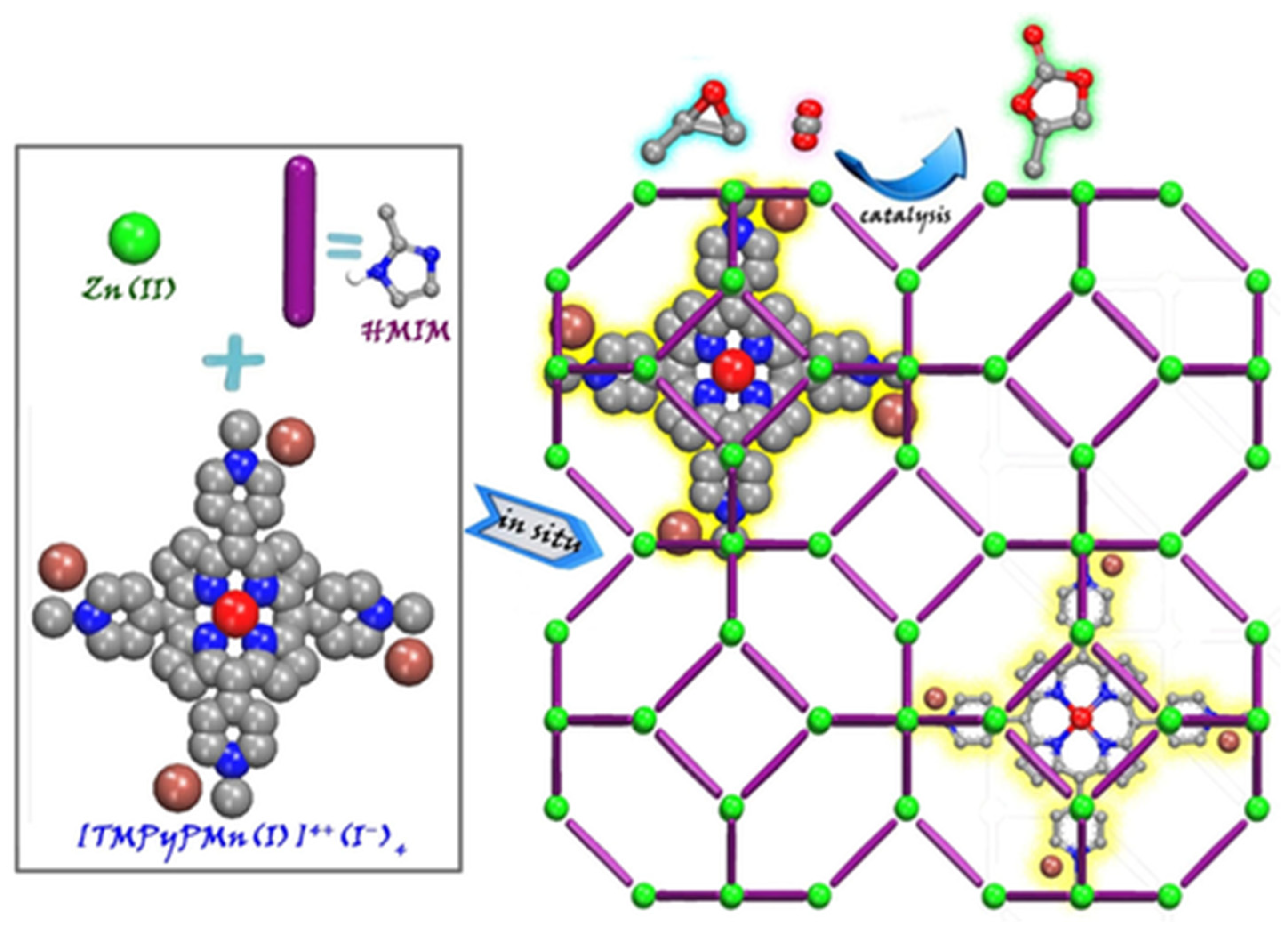
Figure 4. Illustrative demonstration of in situ enveloping of metalloporphyrin into ZIF-8 to conjoin CO2 to epoxide. Reprinted with permission from [48].
2.2. Postsynthetic Procedure of Porph@MOFs Fabrication
For the postsynthetic fabrication of functionalized porph@MOFs, they can be either physically absorbed on the exterior or captured into the cavities, and a specific MOF must be prepared in advance; subsequently, the previously formed porphyrins are incorporated to or grafted on the MOF structure. Next, the metal can be exchanged in porphyrin via controlled-immersion of the final material in a solution of metallic salts. A post-synthetic modification (PSM) of a porphyrin-engulfed MOF to enhance the selective adsorption of CO2 over CH4 was reported [49]. The trapped porphyrin used as a structure-directing agent to provide a “ship-in-a-bottle” mode led to template-based Cd-porph@MOM-11 (MOM; metal–organic materials). In Figure 5a, the effect of the exchange of some cationic organic guests such as H2ppz2+ with Li+ on the selectivity of the adsorption of H2 over N2 was assessed. Remarkably, the results presented that ppz (1,1′,4′,1″,4″,1″′-quaterphenyl-3,5,3″′,5″′-tetracarboxylate) demonstrated significant kinetic trap for both the N2 and H2 ads-des process whilst Li displayed an increment in the pore volume size and more importantly a relatively higher H2 isosteric adsorption heat [50]. In another case, noncatenated hydroxyl-substituted MOF were introduced and replaced by Li+ and Mg2+ ions to convert pendant alcohol to metal alkoxides in order to upgrade the H2 uptake reversibly (Figure 5b). Exchanging was performed via the immersion of as-fabricated MOF in THF solvent (Tetrahydrofuran) to replace the primarily occupied solvent DMF (N-N-Dimethylformamide). Afterwards, the stirring of the respective MOF in an excess of Li+[O(CH3)3−] in CH3 CN/THF solvents was performed to exchange Li+ ions which boosted the hydrogen adsorption ability of the MOF significantly [51]. The illustration in Figure 5c [52] indicates that the MOF of the formula Zn2(NDC)2(diPyNI) (NDC = 2,6-dicarboxylate, diPyNI = N,N′-di-(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide) was reduced by Li0. The interaction imposed by H2 − Li+ inside MOF pores improved its competence to adsorb H2 which was most likely increased by the augmented ligand polarizability and framework displacement. Furthermore, the experimental work in [49] with some modifications of a combination of the first two methods mentioned in Figure 5d submerged single crystals of the prefabricated Cd-porph@MOM-11 into metal chloride salt solutions, with meso-tetra(N-methyl-4-pyridyl) porphine tetratosylate (TMPyP) in methanol serving as a template for PSM, and formed a basis for MOF formation via single-crystal-to-single-crystal ion exchange processes.
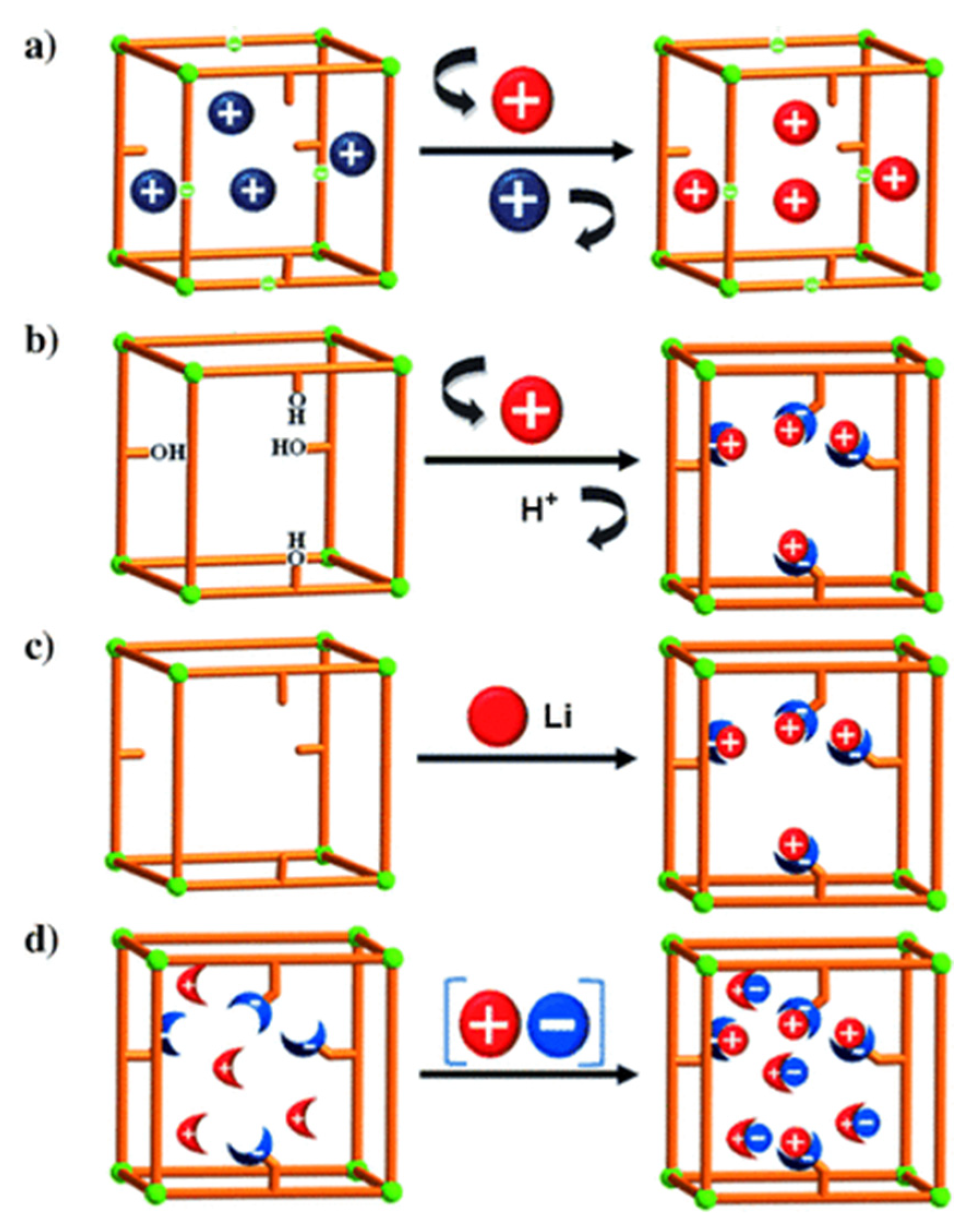
Figure 5. Three basic ways of introduction of open metal sites by PSM synthetic routes to MOFs: (a) cationic guests or organic cations exchange (blue balls) with metal cations (red balls); (b) replacement of a hydroxy protons with Li+ and Mg2+ ions (red balls); (c) chemical reduction of MOM with Li (red balls) and (d) fourth method is a combination of the first two—a collaborative attachment of metal (red balls) chloride (blue ones) salts to anion and cation binding sites. Besides, the sticks and the crescent-shaped bowls attached to sticks are porphyrin-encapsulated inside MOM-11 and cation/anion binding sites. Reprinted with permission from [49].
2.3. Porphyrinic-Oriented MOFs
With regard to porphyrinic MOFs, porphyrin or metalloporphyrin functioning as an organic linker is one of the main components in the framework, which coordinates with secondary building units (SBUs). Accordingly, the choice of suitable shape, size and geometry that meet the desired pore structure to load substrate molecules and to catalyze several reactions efficiently on the surface depends entirely on the rational selection of porphyrins and SBUs. While the insertion of (metallo-) porphyrins not only equips MOFs with new functionality, they can also maintain or bring about far better stability and diversity across the building blocks. As a consequence, the catalytic performances of these types of porous coordination networks could be simply upgraded by regulating Lewis-acid metal active sites and designing well-qualified circumferential functionalities on metalloporphyrins [45]. In line with the previous statements, various parameters such as temperature, solvent, reaction time and the method as well as proper metal nodes and porphyrins chosen, could additionally determine the final product [14].
By applying peripherally functionalized porphyrin/metalloporphyrin as spacer or multidentate ligands directly with metal ions/clusters could lead to the formation of porphyrinic MOFs. Displayed clearly in Figure 6, the porphyrinic MOF PCN-222/MOF-545 (free base-H4TCPP and [Zr6(μ3-O)8(O)8]8− node) was used to selectively oxidize 2-chloroethyl ethyl sulfide (CEES) to a less toxic 2-chloroethyl ethyl sulfoxide (CEESO) at room temperature and neutral pH. The photooxidation of this mustard-gas simulant under mild conditions by exploiting these porous materials as photosensitizer within a half-life of up to 13 min was found to be one of the most convenient methods for the detoxification of such a poisonous compound [53].
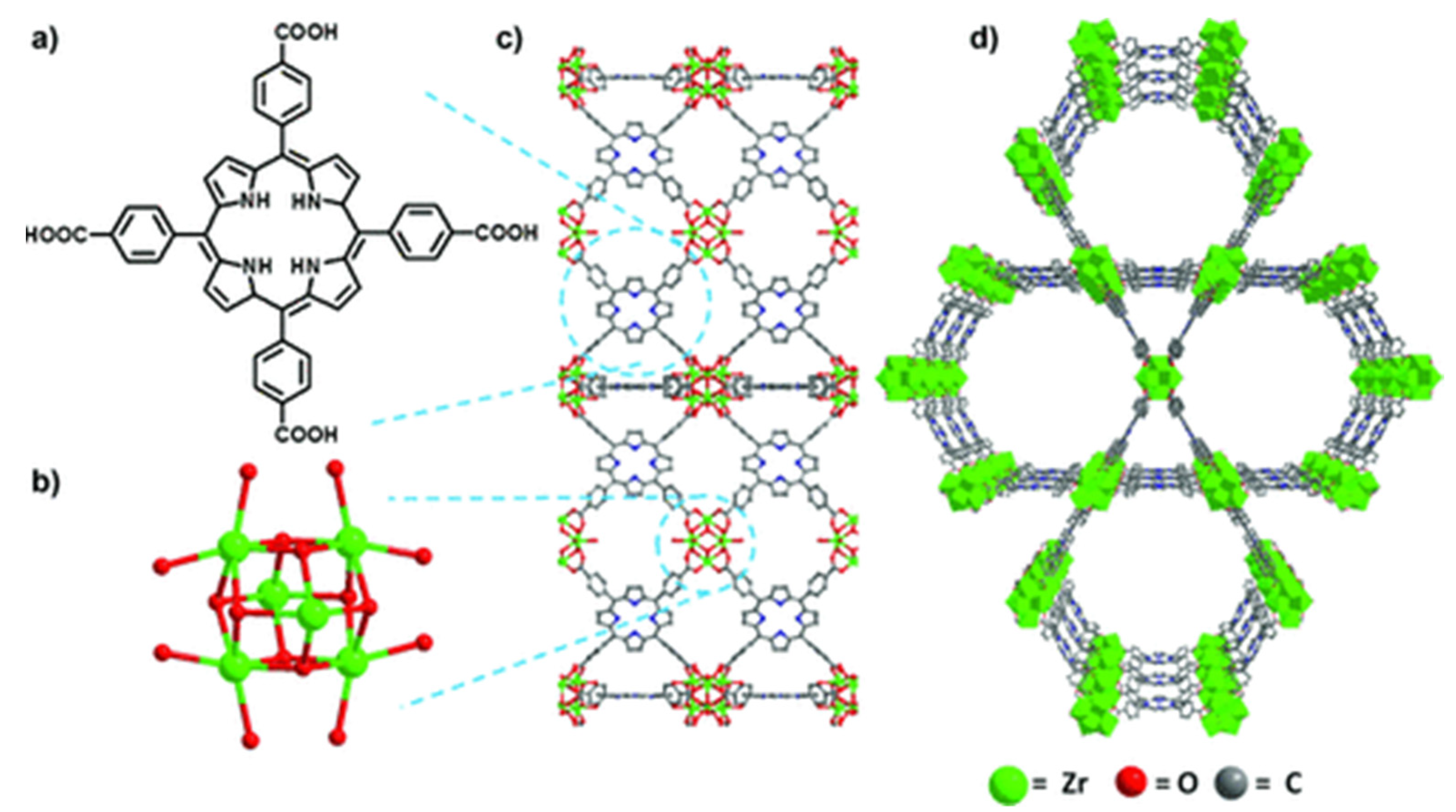
Figure 6. Comparison of free-base PCN-222/MOF-545 (fb-1). (a) Tetrakis(4-carboxyphenyl)porphyrin linker, H4TCPP. (b) [Zr6(m3-O)8(O)8]8¢ node. (c) MOF fb-1, shown across the axis a (d) 3D structure of fb-1, depicted along the c axis. For more clarity hydrogen atoms has been omitted. Reprinted with permission from [53].
Facile insertion of H3PW12O40 inside the solvothermally prefabricated free-base PCN-222 MOF was investigated for the photocatalytic synthesis of some bioactive N-heterocycles such as Nifedipine, Nicardipine, Nicotinic acid (Vitamin B3), and Pyridoxine (Vitamin B6) under visible-LED light irradiation [54]. The porphyrinic Zr-MOF scaffold was constructed by employing TFA and BA as modifiers followed by post-modification with POM to construct the POM@PCN-222 composite.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27154917
References
- Fernandez-Bartolome, E.; Santos, J.; Khodabakhshi, S.; McCormick, L.J.; Teat, S.J.; Saenz de Pipaon, C.; Galan-Mascarós, J.R.; Martín, N.; Costa, J.S. A robust and unique iron (II) mosaic-like MOF. Chem. Commun. 2018, 54, 5526–5529.
- Zaamani, S.; Abbasi, A.; Masteri-Farahani, M.; Rayati, S. One-pot, facile synthesis and fast separation of a UiO-66 composite by a metalloporphyrin using nanomagnetic materials for oxidation of olefins and allylic alcohols. New J. Chem. 2022, 46, 654–662.
- Zhang, K.; Goswami, S.; Noh, H.; Lu, Z.; Sheridan, T.; Duan, J.; Dong, W.; Hupp, J.T. An Iron-Porphyrin Grafted Metal–Organic Framework as a Heterogeneous Catalyst for the Photochemical Reduction of CO2. J. Photochem. Photobiol. 2022, 10, 100111.
- Gutov, O.V.; Bury, W.; Gomez-Gualdron, D.A.; Krungleviciute, V.; Fairen-Jimenez, D.; Mondloch, J.E.; Sarjeant, A.A.; Al-Juaid, S.S.; Snurr, R.Q.; Hupp, J.T.; et al. Water-Stable Zirconium-Based Metal–Organic Framework Material with High-Surface Area and Gas-Storage Capacities. Chem. Eur. J. 2014, 20, 12389–12393.
- Casas-Solvas, J.M.; Vargas-Berenguel, A. Porous Metal–Organic Framework Nanoparticles. Nanomaterials 2022, 12, 527.
- Wu, Z.; Hou, L.; Li, W.; Chen, Q.; Jin, C.; Chen, Y.; Wei, Q.; Yang, H.; Jiang, Y.; Tang, D. Application of a novel biomimetic double-ligand zirconium-based metal organic framework in environmental restoration and energy conversion. J. Colloid Interface Sci. 2022, 610, 136–151.
- Liu, G.; Cui, H.; Wang, S.; Zhang, L.; Su, C.-Y. A series of highly stable porphyrinic metal–organic frameworks based on iron–oxo chain clusters: Design, synthesis and biomimetic catalysis. J. Mater. Chem. A 2020, 8, 8376–8382.
- Min, H.; Wang, J.; Qi, Y.; Zhang, Y.; Han, X.; Xu, Y.; Xu, J.; Li, Y.; Chen, L.; Cheng, K.; et al. Biomimetic Metal–Organic Framework Nanoparticles for Cooperative Combination of Antiangiogenesis and Photodynamic Therapy for Enhanced Efficacy. Adv. Mater. 2019, 31, 1808200–1808210.
- Gharehdaghi, Z.; Rahimi, R.; Naghib, S.M.; Molaabasi, F. Fabrication and application of copper metal–organic frameworks as nanocarriers for pH-responsive anticancer drug delivery. J. Iran. Chem. Soc. 2022, 19, 2227–2737.
- Wang, Y.-C.; Chen, Y.-C.; Chuang, W.-S.; Li, J.-H.; Wang, Y.-S.; Chuang, C.-H.; Chen, C.-Y.; Kung, C.-W. Pore-Confined Silver Nanoparticles in a Porphyrinic Metal–Organic Framework for Electrochemical Nitrite Detection. ACS Appl. Nano Mater. 2020, 3, 9440–9448.
- Lin, W.; Hu, Q.; Jiang, K.; Yang, Y.; Yang, Y.; Cui, Y.; Qian, G. A porphyrin-based metal–organic framework as a pH-responsive drug carrier. J. Solid State Chem. 2016, 237, 307–312.
- Rasheed, T.; Rizwan, K. Metal-organic frameworks-based hybrid nanocomposites as state-of–the-art analytical tools for electrochemical sensing applications. Biosens. Bioelectron. 2022, 199, 113867–113878.
- Sulaiman, M.R.; Gupta, R.K. Nanomaterials for Electrocatalysis (Micro and Nano Technologies), 1st ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 111–144.
- Chen, J.; Zhu, Y.; Kaskel, S. Porphyrin-Based Metal–Organic Frameworks for Biomedical Applications. Angew. Chem. Int. Ed. 2021, 60, 5010–5035.
- Zhang, X.; Wasson, M.C.; Shayan, M.; Berdichevsky, E.K.; Ricardo-Noordberg, J.; Singh, Z.; Papazyan, E.K.; Castro, A.J.; Marino, P.; Ajoyan, Z.; et al. A historical perspective on porphyrin-based metal–organic frameworks and their applications. Coord. Chem. Rev. 2021, 429, 213615–213686.
- Pereira, C.F.; Simões, M.Q.; Tomé, J.P.C.; Paz, F.A.A. Porphyrin-Based Metal-Organic Frameworks as Heterogeneous Catalysts in Oxidation Reactions. Molecules 2016, 21, 1348.
- Abednatanzi, S.; Derakhshandeh, P.G.; Depauw, H.; Coudert, F.-X.; Vrielinck, H.; Van Der Voort, P.; Leus, K. Mixed-metal metal–organic frameworks. Chem. Soc. Rev. 2019, 48, 2535–2565.
- Ohmura, T.; Setoyama, N.; Mukae, Y.; Usuki, A.; Senda, S.; Matsumoto, T.; Tatsumi, K. Supramolecular porphyrin-based metal–organic frameworks: Cu(II) naphthoate–Cu(II) tetrapyridyl porphine structures exhibiting selective CO2/N2 separation. CrystEngComm 2017, 19, 5173–5177.
- Beyzavi, H.; Vermeulen, N.A.; Howarth, A.J.; Tussupbayev, S.; League, A.B.; Schweitzer, N.M.; Gallagher, J.R.; Platero-Prats, A.E.; Hafezi, N.; Sarjeant, A.A.; et al. A Hafnium-Based Metal–Organic Framework as a Nature-Inspired Tandem Reaction Catalyst. J. Am. Chem. Soc. 2015, 137, 13624–13631.
- Shao, S.; Rajendiran, V.; Lovell, J.F. Metalloporphyrin nanoparticles: Coordinating diverse theranostic functions. Coord. Chem. Rev. 2019, 379, 99–120.
- Aziz, A.; Ruiz-Salvador, A.R.; Hernández, N.C.; Calero, S.; Hamad, S.; Grau-Crespo, R. Porphyrin-based metal-organic frameworks for solar fuel synthesis photocatalysis: Band gap tuning via iron substitutions. J. Mater. Chem. A 2017, 5, 11894–11904.
- Zhou, Y.; Zheng, L.; Yang, D.; Yang, H.; Lu, Q.; Zhang, Q.; Gu, L.; Wang, X. Enhancing CO2 Electrocatalysis on 2D Porphyrin-Based Metal–Organic Framework Nanosheets Coupled with Visible-Light. Small Methods 2021, 5, 2000991–2000999.
- Aghayan, M.; Mahmoudi, A.; Nazari, K.; Dehghanpour, S.; Sohrabi, S.; Sazegar, M.R.; Mohammadian-Tabrizi, N. Fe(III) porphyrin metal–organic framework as an artificial enzyme mimics and its application in biosensing of glucose and H2O2. J. Porous Mater. 2019, 26, 1507–1521.
- Rabiee, N.; Rabiee, M.; Sojdeh, S.; Fatahi, Y.; Dinarvand, R.; Safarkhani, M.; Ahmadi, S.; Daneshgar, H.; Radmanesh, F.; Maghsoudi, S.; et al. Porphyrin Molecules Decorated on Metal-Organic Frameworks for Multi-Functional Biomedical Applications. Biomolecules 2021, 11, 1714.
- Sakamaki, Y.; Ozdemir, J.; Heidrick, Z.; Azzun, A.; Watson, O.; Tsuji, M.; Salmon, C.; Sinha, A.; Batta-Mpouma, J.; McConnell, Z.; et al. A Bioconjugated Chlorin-Based Metal–Organic Framework for Targeted Photodynamic Therapy of Triple Negative Breast and Pancreatic Cancers. ACS Appl. Bio Mater. 2021, 4, 1432–1440.
- Liu, W.; Wang, Y.-M.; Li, Y.-H.; Cai, S.-J.; Yin, X.-B.; He, X.-W.; Zhang, Y.-K. Fluorescent Imaging-Guided Chemotherapy-and-Photodynamic Dual Therapy with Nanoscale Porphyrin Metal–Organic Framework. Small 2017, 13, 1603459–1603466.
- Kaim, W.; Schwederski, B.; Klein, A. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life: An Introduction and Guide, 2nd ed.; Wiley: Wiesbaden, Germany, 2013.
- Byrn, M.P.; Curtis, C.J.; Hsiou, Y.; Khan, S.I.; Sawin, P.A.; Tendick, S.K.; Terzis, A.; Strouse, C.E. Porphyrin sponges: Conservative of host structure in over 200 porphyrin-based lattice clathrates. J. Am. Chem. Soc. 1993, 115, 9480–9497.
- Krishna, K.R.; Balasubramanian, S.; Goldberg, I. Supramolecular Multiporphyrin Architecture. Coordination Polymers and Open Networks in Crystals of Tetrakis(4-cyanophenyl)- and Tetrakis(4-nitrophenyl)metalloporphyrin. Inorg. Chem. 1998, 37, 541–552.
- Byrn, M.P.; Curtis, C.J.; Goldberg, I.; Hsiou, Y.; Khan, S.I.; Sawin, P.A.; Tendick, S.K.; Strouse, C.E. Porphyrin sponges: Structural systematics of the host lattice. J. Am. Chem. Soc. 1991, 113, 6549–6557.
- Byrn, M.P.; Curtis, C.J.; Khan, S.I.; Sawin, P.A.; Tsurumi, R.; Strouse, C.E. Tetraarylporphyrin sponges. Composition, structural systematics, and applications of a large class of programmable lattice clathrates. J. Am. Chem. Soc. 1990, 112, 1865–1874.
- Scheidt, R.W. Explorations in metalloporphyrin stereochemistry, physical properties and beyond. J. Porphyr. Phthalocyanines 2008, 12, 979–992.
- Arnold, J. The first structurally characterized alkali metal porphyrin: 7Li NMR behaviour and X-ray crystal structure of the dilithium salt of octaethylporphyrin(2–). J. Chem. Soc. Chem. Commun. 1990, 976–978.
- Scheidt, R.W.; Lee, Y.J. Recent advances in the stereochemistry of metallotetrapyrroles. In Metal Complexes with Tetrapyrrole Ligands I. Structure and Bonding; Buchler, J.W., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 64, pp. 1–70.
- Goldberg, I. Crystal engineering of porphyrin framework solids. Chem. Commun. 2005, 1243–1254.
- Krishna, K.R.; Balasubramanian, S.; Goldberg, I. Self-Assembly of Functionalized Metalloporphyrins into Microporous Polymeric Networks. Mol. Cryst. Liq. Cryst. 1998, 313, 105–114.
- Sato, T.; Mori, W.; Kato, C.N.; Ohmura, T.; Sato, T.; Yokoyama, K.; Takamizawa, S.; Naito, S. Microporous rhodium (II) 4,4′,4″,4‴- (21H,23H-porphine-5,10,15,20-tetrayl) teteakisbenzoate, synthesis, nitrogen adsorption properties and catalytic performance for hydrogenation of olefin. Chem. Lett. 2003, 32, 854–855.
- Sato, T.; Mori, W.; Kato, C.N.; Yanaoka, E.; Kuribayashi, T.; Ohtera, R.; Shiraishi, Z. Novel microporous rhodium(II) carboxylate polymer complexes containing metalloporphyrin: Syntheses and catalytic performances in hydrogenation of olefins. J. Catal. 2005, 232, 186–198.
- Longevial, J.-F.; Clément, S.; Wytko, J.A.; Ruppert, R.; Weiss, J.; Richeter, S. Peripherally Metalated Porphyrins with Applications in Catalysis, Molecular Electronics and Biomedicine. Chem. Eur. J. 2018, 24, 15442–15460.
- Feng, L.; Wang, K.-Y.; Joseph, E.; Zhou, H.-C. Catalytic Porphyrin Framework Compounds. Trends Chem. 2020, 2, 555–568.
- Masih, D.; Chernikova, V.; Shekhah, O.; Eddaoudi, M.; Mohammed, O.F. Zeolite-like Metal–Organic Framework (MOF) Encaged Pt(II)-Porphyrin for Anion-Selective Sensing. ACS Appl. Mater. Interfaces 2018, 10, 11399–11405.
- Wang, K.; Lv, X.-L.; Feng, D.; Li, J.; Chen, S.; Sun, J.; Song, L.; Xie, Y.; Li, J.-R.; Zhou, H.-C. Pyrazolate-Based Porphyrinic Metal–Organic Framework with Extraordinary Base-Resistance. J. Am. Chem. Soc. 2016, 138, 914–919.
- Yamazaki, S.I. Metalloporphyrins and related metallomacrocycles as electrocatalysts for use in polymer electrcolyte fuel cells and water electrolyzers. Coord. Chem. Rev. 2018, 373, 148–166.
- Gottfried, J.M. Surface chemistry of porphyrins and phthalocyanines. Surf. Sci. Rep. 2015, 70, 259–379.
- Chakraborty, J.; Nath, I.; Verpoort, F. Snapshots of encapsulated porphyrins and heme enzymes in metal-organic materials: A prevailing paradigm of heme mimicry. Coord. Chem. Rev. 2016, 326, 135–163.
- Cai, H.; Huang, Y.-L.; Li, D. Biological metal–organic frameworks: Structures, host–guest chemistry and bio-applications. Coord. Chem. Rev. 2019, 378, 207–221.
- Yan, T.; Guo, J.-H.; Liu, Z.-Q.; Sun, W.-Y. Metalloporphyrin Encapsulation for Enhanced Conversion of CO2 to C2H4. ACS Appl. Mater. Interfaces 2021, 13, 25937–25945.
- He, H.; Zhu, Q.-Q.; Zhang, C.; Yan, Y.; Yuan, J.; Chen, J.; Li, C.-P.; Du, M. Encapsulation of an Ionic Metalloporphyrin into a Zeolite Imidazolate Framework in situ for CO2 Chemical Transformation via Host-Guest Synergistic Catalysis. Chem. Asian J. 2019, 14, 958–962.
- Zhang, Z.; Gao, W.-Y.; Wojtas, L.; Ma, S.; Eddaoudi, M.; Zaworotko, M.J. Post-Synthetic Modification of Porphyrin-Encapsulating Metal–Organic Materials by Cooperative Addition of Inorganic Salts to Enhance CO2/CH4 Selectivity. Angew. Chem. Int. Ed. 2012, 51, 9330–9334.
- Yang, S.; Lin, X.; Blake, A.J.; Walker, G.S.; Hubberstey, P.; Champness, N.R.; Schröder, M. Cation-induced kinetic trapping and enhanced hydrogen adsorption in a modulated anionic metal–organic framework. Nat. Chem. 2009, 1, 487–493.
- Mulfort, K.L.; Farha, O.K.; Stern, C.L.; Sarjeant, A.A.; Hupp, J.T. Post-Synthesis Alkoxide Formation Within Metal−Organic Framework Materials: A Strategy for Incorporating Highly Coordinatively Unsaturated Metal Ions. J. Am. Chem. Soc. 2009, 131, 3866–3868.
- Mulfort, K.L.; Hupp, J.T. Chemical Reduction of Metal−Organic Framework Materials as a Method to Enhance Gas Uptake and Binding. J. Am. Chem. Soc. 2007, 129, 9604–9605.
- Liu, Y.; Howarth, A.J.; Hupp, J.T.; Farha, O.K. Selective Photooxidation of a Mustard-Gas Simulant Catalyzed by a Porphyrinic Metal–Organic Framework. Angew. Chem. Int. Ed. 2015, 127, 9129–9133.
- Karamzadeh, S.; Sanchooli, E.; Oveisi, A.R.; Daliran, S.; Luque, R. Visible-LED-light-driven photocatalytic synthesis of N-heterocycles mediated by a polyoxometalate-containing mesoporous zirconium metal-organic framework. Appl. Catal. B 2022, 303, 120815.
This entry is offline, you can click here to edit this entry!