Inflammation represents a main feature of chronic liver diseases and plays a key role in any stages of the fibrogenic process, even during fibrosis regression. Inflammatory response involves multicellular interactions, dynamically regulated by a plethora of factors (e.g., soluble mediators, ECM components, pathogen-associated molecular patterns-PAMPs, damage-associated molecular patterns-DAMPs), acting in cell-specific fashion and aimed to restore liver architecture and function, but also leading to liver fibrosis when the noxious agent persists.
Cell death is an early and primary inducer of chronic inflammation and fibrosis. Hepatocyte-derived apoptotic bodies stimulate the secretion of pro-inflammatory and profibrogenic cytokines from macrophages and promote activation of HSCs through induction of autophagy [
100,
101,
102]. In addition, injured hepatocytes release DAMPs, such as ATP, phormyl peptides, High Mobility Group Box 1(HMGB1) [
103] and cytokines such as IL-33 [
104], which triggers HSC activation directly or indirectly, by promoting IL-13 release by innate lymphoid cells (ILC2). At the same time, inflammatory mediators secreted by infiltrating immune cells contribute to cell death, amplifying hepatic injury [
104].
As major effectors of fibrosis, activated HSCs play a central role in inflammation, receiving a wide variety of stimuli from inflammatory cells and from hepatocytes, cholangiocytes and activated sinusoidal endothelial cells (SECs). Activated HSCs are highly responsive to inflammatory mediators which induce inflammatory pathways (such as NF-κB and AP-1) [
105,
106] and consequent secretion of cytokine/chemokines that act in autocrine and paracrine fashion. Inflammatory signals exert specific roles on HSCs, maintaining survival (IL-1β, TNFα, CXCL12) and the activated state (ILs and chemokines) [
107], providing chemotactic stimuli for HSCs themselves or inflammatory cells (CCL2, CCL5, CXCL9, CXCL10, CX3CL1) and mediating the gut-liver axis crosstalk (toll like receptors (TLRs)) [
105]. All these processes can contribute to positively or negatively modulate inflammatory responses and fibrogenesis, promoting fibrosis progression or regression.
As modulators of liver fibrosis, immune cells exhibit a dual role, being able to contribute to both fibrosis progression and regression [
108,
109]. Danger signals generated in the site of injury lead to infiltration of circulating inflammatory cells (T lymphocytes, neutrophils, dendritic cells and monocytes) and activation of Kupffer cells (KCs) [
108,
109]. The release of a wide range of soluble mediators amplifies inflammation and stimulates the fibrogenic process. Upon removal of the cause of injury, the balance switches from pro- to anti-inflammatory/restorative pathways, promoting fibrosis resolution. This shift is achieved by rearrangements in the type of immune cell populations recruited, with a marked drop in intrahepatic T cells and blood-derived cells (NKT cells, monocytes) [
110], and phenotypic modifications of certain cell types, mainly macrophages.
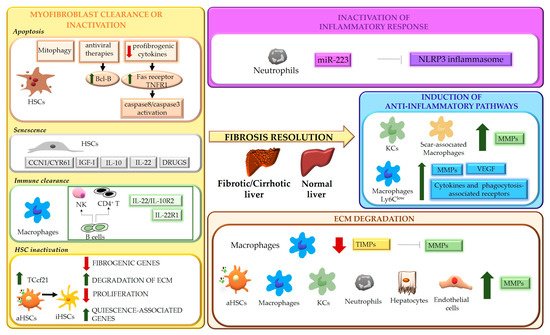
5. ECM Degradation
Liver fibrosis is a dynamic process characterized by an unfavorable balance between ECM deposition and degradation. Degradation of ECM represents one of the most relevant aspects of fibrosis regression and requires activation of MMPs, macrophage phagocytic activity and downregulation of MMP-inhibitory molecules, such as tissue inhibitors of MMPs, TIMPs [
7,
10]. MMPs are the main matrix-degrading enzymes [
62] and, according to substrate specificity, can be grouped in collagenases (MMP8, MMP1 and MMP13) which cleave native fibrillar collagens to gelatin, gelatinases (MMP2, MMP9), degrading a wide range of substrates including gelatin, collagens and, in some extent, elastin, metalloelastases (MMP12) and others (
Table 1).
Table 1. Classification of human metalloproteinases (MMPs) and their function.
|
MMPs
|
GROUP
|
FUNCTION
|
|
MMP1, MMP8, MMP13
|
Collagenases
|
Cleavage of native fibrillar collagens to gelatin
|
|
MMP2, MMP9
|
Gelatinases
|
Degradation of a wide range of substrates, including gelatin, collagens and elastin
|
|
MMP12
|
Metalloelastases
|
Elastin degradation
|
They are secreted by various cell types, including aHSCs, hepatocytes, endothelial cells, and inflammatory cells, such as neutrophils and macrophages. MMP release and activity are finely regulated during the different phases of fibrogenic process, as well as during fibrosis regression. In this context, a relevant role in matrix degradation is played by “restorative” macrophages that, besides a role in phagocytic digestion of matrix fragments, represent a major source of MMP12, MMP13, and MMP9 [
124]. In a recent study, Feng et al. showed that in thioacetamide (TAA)-induced fibrosis, depletion of KCs delayed resolution following toxin withdrawal and this was mainly ascribed to a marked decrease in MMP9 [
135]. Because activated HSCs display high TIMP levels, ECM degradation strictly correlates with HSC clearance and the subsequent shift in MMPs/TIMPs balance, creating the conditions for a milieu favoring parenchymal regeneration.
6. Reversibility of Cirrhosis
ECM remodeling is crucial in determining reversibility of fibrosis. In recent years, clinical and experimental studies have provided evidence that matrix remodeling and at least partial restitution towards a normal architecture may be observed even in advanced liver fibrosis or cirrhosis [
136,
137,
138,
139,
140]. The amount of elastin and cross-linked proteins in fibrotic scars is critical in this process. Protein cross-linking, which is mediated by cellular transglutaminases and lysyl oxidases, stabilizes ECM, enhancing its resistance to enzyme degradation and, together with elastin, increases matrix stiffness, that further sustains HSC activation via integrin-mediated mechanisms [
62]. In this setting, MMP12 released by macrophages can still promote matrix turnover acting not only on elastin but also on collagens [
141]. However, ECM remodeling in cirrhotic scars is also influenced by vascular remodeling that can hamper matrix degradation [
142]. Thus, even when restitution to normal liver architecture is achieved, cirrhosis-associated derangements in the vascular system and in other organs persist. By using two different models of cirrhosis induction and reversal (TAA and BDL), Hsu et al. demonstrated that, despite a complete regression of fibrotic scars, portal hypertension was only partially reduced, due to persisting alterations in splanchnic and collateral circulation [
143].
These biologic considerations have clear clinical implications. Regression of fibrosis represents a major clinical goal, since it can lead to a recovery of liver function and reduction in portal pressure, which decrease the incidence of portal-hypertensive complications and of hepatocellular carcinoma (HCC) [
144,
145,
146,
147]. It is well known that mild and moderate fibrosis can be reversible, but the same concept is not always true for cirrhosis. In this respect, the identification of a “point of no return” in the natural history of liver disease can be very difficult, despite its utmost relevance in clinical practice. This may be viewed as a condition beyond which even causal therapy (e.g., viral eradication) does not determine a significant regression of fibrosis and/or has limited impact on the appearance of complications and prognosis of the patient. As indicated above, the degree and amount of structural damage, in particular the development of extensive matrix crosslinking [
148] and accumulation of elastin fibers in long-standing cirrhosis, have been indicated as a major element to identify the “point of no return” [
141].
From a clinical standpoint, the ‘model’ of HCV eradication has provided relevant data in this context. Patients with compensated cirrhosis (Child-Turcotte-Pugh class A) achieving viral eradication with direct-acting antivirals (DAAs), show regression of fibrosis in a relevant percentage of cases (88%) [
149] and a consequent decrease of portal hypertension [
150]. When patients with cirrhosis Child-Pugh class B and C are considered, long-term data about the effects of sustained virologic response (SVR) after DAA treatment on fibrosis and liver-related complications and survival are less abundant. However, data from other contexts (e.g., HBV or alcohol-related decompensated cirrhosis) indicated that Child C class could represent a “point of no return” in terms of fibrosis decrease even after removal of the etiologic factor [
151,
152]. Other clinical predictors of the lack of fibrosis regression include age (>65 years), albumin (<3.5 g/dL), high MELD score (>20), alcohol habit and presence of metabolic disorders. However, none of them are satisfactory consistent to be used in clinical practice [
153].
Advanced liver fibrosis and cirrhosis are major risk factors for HCC [
154,
155]. In particular, fibrosis and cancer-associated fibroblasts (CAF,) can influence the onset of HCC modulating the cancer microenvironment [
156,
157]. Considering these assumptions, HCV eradication should determine a decrease of both HCC occurrence and recurrence. In recent years, this has been a very debated issue since some studies suggested that SVR due to DAA, differently from interferon-based therapies, could increase the risk of both occurrence and recurrence of liver cancer [
158]. It is now accepted that there is no such risk on a population basis and, as recently demonstrated [
159], SVR due to DAAs leads to a drop in all-cause mortality, hepatic decompensation, and HCC. Nevertheless, on an individual basis, DAAs might favor the HCC development in subjects who already have a predisposing hepatic condition such as activated neo angiogenesis [
160]. Moreover, subjects with severe metabolic impairment may have a risk of HCC despite viral eradication [
161]. DAA-induced modifications in VEGF, epidermal growth factor, and inflammatory factors have been proposed for the detection of subgroups at risk of HCC, and some authors have proposed these as possible determinants of the susceptibility to cancer development [
162,
163].
7. Vascular Remodeling
As anticipated above, angiogenesis and vascular remodeling represent additional mechanisms involved in both fibrosis development and regression. Although the role of angiogenesis in promoting liver fibrosis is fully accepted, new lines of evidence indicate that angiogenic factors may also induce scar degradation and tissue repair during fibrosis resolution. Using murine models of fibrosis reversal, Yang et al. showed that the VEGF/VEGFR2 pathway is essential to maintaining sinusoidal permeability and the subsequent monocyte infiltration and macrophage fibrinolytic activity [
128]. Moreover, VEGF release by macrophages was shown to be critical for fibrosis resolution. In fact, VEGR2-mediated activation by VEGF induced ECM degradation through upregulation of MMPs and downregulation of TIMPs in sinusoidal endothelial cells [
164].
Capillarization of the sinusoids and changes in liver sinusoidal endothelial cells (LSECs) represent key events in liver fibrogenesis, triggering HSC activation and impairing hepatocyte polarization. These consist in LSEC dedifferentiation with loss of fenestrae and deposition of a continuous basement membrane that hampers normal exchanges between blood circulation and hepatocytes. Restoration of differentiated LSEC is crucial for recovery from hepatic fibrosis, as proved by the fact that depleting factors implicated in sinusoidal permeability, such as VEGF or CXCL9, results in delayed recovery [
128]. In a thioacetamide-induced rat model of cirrhosis, administration of BAY 60-2770, an activator of soluble guanylate cyclase (sGC), promoted a complete reversal of sinusoid capillarization, by restoring normal levels of cGMP, fenestrae, and porosity in LSECs. Restitution to differentiated LSECs resulted in reversal of HSC activation and regression of fibrosis. Moreover, maintenance of physiological levels of cGMP in LSECs was essential to prevent fibrosis progression [
165]. Liver X Receptor (LXR) α, which mediates multiple antifibrogenic actions interfering with the activation of HSCs, the release of inflammatory mediators and the synthesis of profibrogenic factors [
56,
166,
167,
168], was hypothesized to play a role in reverting capillarization of the sinusoids, through inhibition of Hedgehog-dependent signaling in LSECs [
169]. In a mouse model of biliary fibrosis induction and reversal, Lee et al. identified AKAP12, a scaffold protein expressed in various cell types regulating cyclic adenosine monophosphate (cAMP) compartmentalization, as a novel mediator of fibrosis resolution, through mechanisms affecting LSEC dedifferentiation/activation and angiogenesis [
170]. In an elegant study, Xu et al. identified leukocyte cell-derived chemotaxin 2 (LECT2) as a ligand of Tie1 (an orphan receptor expressed by endothelial cells) and LECT2-Tie1 as a novel profibrogenic pathway involved in vascular remodeling, that enhances sinusoid capillarization and reduces portal angiogenesis. They showed that knockdown of LECT2 (in both LECT2 KO mice and AAV9-LECT2 shRNA- treated mice) attenuates fibrosis development and ameliorates established fibrosis in different experimental models, reducing sinusoid capillarization and increasing portal angiogenesis. Notably, serum levels of LECT2 were significantly increased in patients with advanced fibrosis, indicating LECT2-Tie1 signaling as a promising therapeutic target [
171].