Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Alzheimer’s Disease (AD) is the most common neurodegenerative disease worldwide, with a high prevalence that is expected to double every 20 years. Besides the formation of Aβ plaques and neurofibrillary tangles, neuroinflammation is one the major phenotypes that worsens AD progression. Indeed, the nuclear factor-κB (NF-κB) is a well-established inflammatory transcription factor that fuels neurodegeneration.
- Alzheimer’s
- drug discovery
- inflammation
- neurodegeneration
- NF-κB
1. The NF-κB Family and Its General Role in Inflammation
The nuclear factor-κB (NF-κB) is composed of a family of five transcription factors involved in various cellular processes, and it is particularly notorious for its role in mediating inflammatory responses. This family consists of NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel. NF-κB activation promotes the transcription of target genes, many of which are proinflammatory. The NF-κB signaling is activated via two distinct pathways: the canonical and noncanonical pathways. Notably, the canonical pathway is highly studied and plays a critical role in inflammatory responses, a key characteristic in AD development. In an inactive state, the p65/p50 dimers in the canonical NF-κB pathway are sequestered in the cytoplasm by IκBα. Upon exposure to proinflammatory stimuli such as cytokines, pathogens, and danger-associated molecular patterns, the p65/p50 dimers are released from IκBα due to the phosphorylation cascade that results in the proteasomal degradation of IκBα. Subsequently, p65/p50 translocates into the nucleus, where it binds to its cognate κB motif, leading to the activation and expression of NF-κB target genes [3]. On the other hand, the non-canonical pathway is activated through a subset of Tumor Necrosis Factor Receptor (TNFR) superfamily members, leading to the activation of NF-κB inducing kinase (NIK). NIK phosphorylates IκB kinase alpha (IKKα), which phosphorylates the C-terminal of p100 to generate p52. Following the phosphorylation cascade, p52/RelB translocates into the nucleus, triggering the expression of NF-κB target genes that play a role in immune cells’ development [4].
As a result of NF-κB’s extensive involvement in cellular inflammatory responses, it has become an attractive target for research on inflammatory diseases. In the AD brain, Toll-like Receptors (TLRs) are overexpressed on microglia and neurons. TLRs mainly activate the canonical NF-κB signaling pathway, leading to the expression of proinflammatory factors [5]. Microglial activation is one of the early events that lead to AD development, given that the primary function of microglia in the brain is the protection from pathogens and the clearance of cellular debris, including amyloid beta (Aβ) plaque formation. Thus, the activation of NF-κB signaling and consequent release of cytokines and chemokines from microglia results in chronic inflammation observed in AD [6]. As such, the role of NF-κB in AD is an important topic that warrants more attention in the field of AD research. Herein, we discuss the contribution of NF-κB signaling to AD pathology and provide an overview of current drugs approved/in development for AD, including NF-κB inhibitors.
2. Role of NF-κB in AD
As aforementioned, neuroinflammation, oxidative stress, and apoptosis are some of the key processes that contribute to the fatality of AD patients. The major histological features essential for AD diagnosis, such as formation of amyloid-beta (Aβ) plaques and neurofibrillary tangles (NFT) in neurons, can be exacerbated by inflammation perpetuated by glial cells [7]. Consequently, Aβ and NFT promotes neuronal loss and instability [8]. NF-κB is central to this vicious cycle of neurodegeneration observed in AD [9]. However, depending on the cell type and/or combination of NF-κB subunits, the activation of NF-κB can play a dual role in either neuroprotection or neurodegeneration [10]. This is evident from several studies that have shown the expression of proapoptotic genes which cause neuronal death via the transactivation of p65/p50 dimers. On the contrary, c-Rel-containing dimers mediate anti-apoptotic gene expression, thereby, promoting neuronal survival. c-Rel or p65/p50 heterodimers can be selectively activated depending on the nature of stimuli received such as IL-1β, Nerve Growth Factor (NGF), Aβ peptide, or glutamate [11,12].
Mechanistically, NF-κB plays a crucial role in AD pathogenesis by regulating different molecules responsible for promoting the morbidities associated with AD. Below, we will provide a synopsis of various representative factors that are involved in the activated NF-κB signaling in AD pathogenesis.
2.1. NF-κB and β-Secretase in AD
Studies have shown increased NF-κB levels in the cerebral cortex of AD patients, and this correlates with high levels of β-site amyloid precursor protein (APP) cleaving enzyme-1 (BACE1). The data from a previous study demonstrate that the p65 subunit of NF-κB binds to the κB elements on the promoter of BACE1, inducing the expression of β-secretase [13]. High levels of β-secretase facilitate the amyloidogenic pathway of APP processing, resulting in the formation of amyloid fibrils, which consequently aggregate into amyloid plaques (Figure 1) [14]. Similarly, Aβ oligomers can in turn stimulate NF-κB activation in neurons and glial cells [15]. Aβ40 peptide was shown to strongly activate the p65/p50 dimers of NF-κB and induced the expression of pro-apoptotic genes in primary and N2TN neurons. Some of the genes expressed following Aβ40 induction include Bax, p63, DcR2, and TANK (TRAF family member-associated NF-κB activator), and they all possess κB regulatory elements in their promoter region. Also, Aβ40 increased the accumulation of Aβ42 aggregates, further promoting the neuropathology cascade of AD [16]. Similarly, Aβ (25–35) peptide was shown to cause toxicity in primary neurons and cell lines through increased production of peroxides, a source of oxidative stress. This phenomenon is also accompanied by high levels of NF-κB signaling [17]. Because reactive oxygen species (ROS) are known to activate NF-κB subunits in some cases, this study suggests an indirect link between Aβ peptide-mediated toxicity and NF-κB activation.
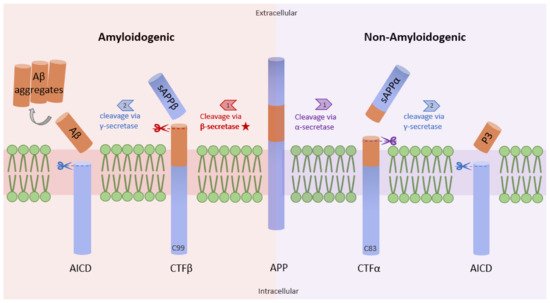
Figure 1. Amyloidogenic and non-amyloidogenic pathways. APP (center) can undergo proteolytic processing through two unique pathways, amyloidogenic processing (left) and non-amyloidogenic processing (right). In amyloidogenic processing, β-secretase cleaves APP, forming C99 and sAPPβ. C99 is further cleaved by γ-secretase to form amyloid beta peptides (Aβ). Importantly, the Aβ formation rate is dependent on the cleavage rate of APP by β-secretase. In non-amyloidogenic processing, APP is cleaved by α-secretase to form C83 and sAPPα, which can be further cleaved by γ-secretase, yielding p3 (adapted from [14]).
2.2. NF-κB in Reactive Microglia and Astrocytes
Considering the role of glial cells in inflammation, NF-κB signaling in reactive microglia and astrocytes has been reported to contribute to AD pathology. Bacteroides fragilis Lipopolysaccharide (BF-LPS) activated the NF-κB pathway via distinct Toll-like receptors (TLR2 and TLR4) and CD14 receptors present on microglial cell surfaces in a neuron-glial co-culture experiment. BF-LPS, among other several activators of NF-κB such as Aβ42, TNF-α, and IL-1β, exhibited the highest potency in p65/p50 dimer activation [18]. Another study demonstrated the high levels and co-localization of LPS with Aβ42. LPS was shown to promote the NF-κB-dependent transcription of cytokines, causing increased accumulation of Aβ plaques and increased tau protein hyperphosphorylation. The generation of high levels of proinflammatory cytokines, such as IL-1, IL-6, and TNF-α, damages the oligodendrocytes, promoting myelin injury [19]. This leaves the neurons vulnerable to Aβ neurotoxicity and promotes the autocrine loop required for AD progression [19]. Astrocytes, a macroglial cell, which is pivotal to maintaining brain homeostasis, also promote neurodegeneration via NF-κB, leading to Aβ42 accumulation, pro-inflammatory cytokine production, and the generation of inducible Nitric Oxide Synthase (iNOS) [20,21,22].
2.3. NF-κB and ApoE in AD
Additionally, apolipoprotein E allelic variants are associated with AD development, with APOEε2 causing reduced risk and APOEε4 causing increased risk in comparison to the common allele, APOEε3. Particularly, APOEε4 was discovered to inhibit Aβ clearance from the brain interstitial fluid through various mechanisms [23]. In another study, it was demonstrated that Aβ40 could activate NF-κB and lead to the increase of APOE promoter function. The regulatory region of the APOE gene has been further characterized and was shown to contain two functional κB motifs. In this study, the NF-κB inhibitor sodium salicylate was further applied to evaluate the effect of NF-κB inhibition on the promoter activity of APOE in AD. Data suggested that NF-κB-dependent APOE promoter activity was significantly decreased upon the treatment with NF-κB inhibitor [24]. Thus, considering that APOE enhances Aβ fibril formation in AD pathogenesis, the use of an NF-κB inhibitor may lessen NF-κB-induced APOE activity in the AD brain. Similarly, gene clustering analysis by Ophir et al. revealed the greater activation of NF-κB and upregulation of NF-κB-inducible genes in APOE4 mice when compared to APOE3 mice following treatment with LPS. These upregulated genes include chemokines and inflammatory cytokines like IL-1β, IL-6, CCL-3, and TNF-α, among others [25].
2.4. NF-κB and Glutamate in AD
Furthermore, NF-κB is involved in Aβ oligomer-induced glutamate excitotoxicity which contributes to the AD neurodegeneration cascade. Aβ peptides have been shown to increase glutamate receptor activation with concomitant increases in intracellular calcium levels in human cerebral cortical neurons. Sustained increases in the levels of intracellular calcium is known to cause microtubule instability, increased tau phosphorylation via calcium dependent kinases, mitochondrial oxidation impairment, and ultimately increased ROS generation [26,27]. Notably, Lim and colleagues confirmed high levels of calcium and metabotropic glutamate receptor 5 (mGluR5) near Aβ plaques in the hippocampal astrocytes of AD patients. This study reveals that Aβ42 increases cytosolic calcium levels by activating calcineurin (CaN), which in turn enables the NF-κB-dependent transcription of mGluR5. It was shown that the activation of NF-κB by CaN might have occurred via the CaN dephosphorylation of B-cell lymphoma 10 (Bcl10) [28]. Bcl10 is known to activate the NF-κB pathway via ubiquitination of IKK-γ [29]. Similarly, mGluR5 staining co-localizes with the accumulated nuclear p65 subunit of NF-κB in hippocampal astrocytes, further reinforcing the link between NF-κB and glutamate in promoting AD-like pathology [28].
2.5. NF-κB and miRNAs in AD
Beyond the aforementioned factors, NF-κB also exerts its neurotoxic effect in AD via the regulation of microRNA. MicroRNAs such as miRNA-125b, miRNA-9, miRNA-155, miRNA-34a, miRNA-146a have been shown to be regulated by NF-κB [30]. Notably, miRNA-125b is the most abundant in the human brain and is highly upregulated in AD tissues [31]. NF-κB-activated mir-125b was reported to inhibit complement factor H (CFH) in human neuronal-glia cells. CFH is known to play an important role in suppressing the innate immune system by inhibiting the conversion of C3 to C3b in the complement pathway [32]. Similarly, NF-κB-induced mir-125b has been shown to silence 15-lipoxygense (15-LOX) expression, an enzyme that is important for the conversion of docohexaneoic acid to neuroprotectin D1 (NPD1) and vitamin D3 receptor (VDR). Both NPD1 and VDR are essential for protecting the brain from the toxic effects of ROS and reactive nitrogen species (RNS) [31]. Another study showed that miRNA-34a downregulates Triggering Receptor Expressed in Myeloid Cells 2 (TREM2) in the hippocampal CA1 region of AD patients. TREM2 plays a crucial role in the microglial clearance of Aβ [33]. Taken together, the aforementioned evidence suggests the expansive role of NF-κB in AD progression through the regulation of microRNA expression.
2.6. NF-κB and Tau Pathology in AD
Additionally, NF-κB signaling contributes to tau pathology. A study demonstrated that NF-κB can induce the expression of SET gene isoform 1, which is upregulated in AD patients’ brains [34]. SET was shown to contain a functional κB sequence in its promoter region. In the cytoplasm, SET causes the inhibition of protein phosphatase 2A (PP2A), a major tau phosphatase that prevents tau hyperphosphorylation. SET is also known to bind to the pro-apoptotic domain of APP, leading to neuronal apoptosis [34]. Comparably, a previous study observed the glycosylation of paired helical filament tau by advanced glycation end products (AGE). AGE-tau was shown to generate high levels of ROS, resulting in nuclear translocation of p65/p50 dimers and consequently increased IL-6, APP, and Aβ production in primary cortical neurons and neuroblastoma cells [35].
To sum up, these aforementioned examples of NF-κB signaling and its regulation of various gene expressions in neuronal and glial cells underscores the role of NF-κB in perpetuating a cycle of neurodegeneration in AD (Figure 2).
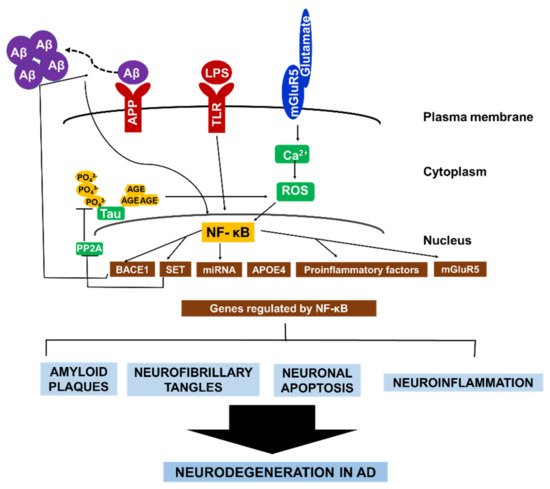
Figure 2. The central role of NF-κB in AD pathology. NF-κB facilitates the autocrine production of several factors that constitute AD pathology. NF-κB activates BACE1, which promotes Aβ fibril formation. Consequently, Aβ fibrils directly activate NF-κB, leading to the expression of APOE4, and mGluR5. In microglia and astrocytes. Aβ42 activates NF-κB, which induces the expression of proinflammatory factors that causes myelin injury. Additionally, NF-κB activates microRNA production which suppresses the expression of various neuroprotective factors. Similarly, the formation of hyperphosphorylated tau in AD brain is enhanced by NF-κB-dependent activation of SET, which inhibit tau’s dephosphorylation. Conversely, glycated tau triggers ROS production, leading to NF-κB activation. Collectively, the different pathways that contribute to neurodegeneration in AD are highly interconnected via continuous NF-κB activity in both neurons and glial cells.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23168972
This entry is offline, you can click here to edit this entry!