Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Learning and memory formation rely on the precise spatiotemporal regulation of gene expression, such as microRNA (miRNA)-associated silencing, to fine-tune gene expression for the induction and maintenance of synaptic plasticity. Here, we summarize studies that have manipulated miRNA expression using various approaches in rodents, with changes in cognitive performance. We also summarize extensive studies on miRNAs correlated with pathogenic tau and amyloid-β that drive the processes of Alzheimer’s disease (AD).
- learning impairment
- microRNA
- memory impairment
1. Direct Evidence of miRNA Regulation in Learning and Memory
Brain-specific or brain-enriched miRNAs, some of which are transcriptionally induced or promote the turnover of mature forms by neuronal activity, are widely expressed in different brain regions [17]. It is now well established that the biogenesis, activity, and degradation of specific miRNAs regulate neuronal plasticity, which is responsible for many complex brain functions, including the learning/memory process, and that the misexpression of some miRNAs is associated with neurological disorders [18,19]. For example, the overexpression of miR-34a in neurons was found to negatively affect dendritic growth and arborization and weakened synaptic plasticity by reducing synaptotagmin-1 expression in cortical neuronal cultures [20]. In animal experiments, auditory fear conditioning upregulated miR-34a in the basolateral amygdala, whereas antagonizing miR-34a by miRNA sponges suppressed auditory fear memory [21]. Virus-mediated overexpression of miR-34a in the lateral ventricle enhanced MWM performance by promoting neural progenitor proliferation [22]. However, transgenic overexpression of miR-34a in the whole brain exhibited profound behavioral impairment in the T-maze task, accompanied by accumulation of intracellular Aβ and tau hyperphosphorylation [23]. Overexpression of miR-34c by the injection of mimics or lentivirus into the hippocampus of wild-type (WT) mouse brain impaired learning and memory formation in multiple behavioral experiments, including MWM, CFC, and NOR. However, the expression of miR-34c was shown to increase with age in mice and in humans with the onset of AD, contributing to impairments in CFC memory. When miR-34c inhibitors were injected into the hippocampus, the memory-impairing phenotypes were rescued in an aged AD mouse model (APP/PS1-21) by targeting Sirtuin 1 (SIRT1) [24]. Similar results were observed in another AD mouse model (SAMP8), indicating that miR-34c mediates synaptic and memory deficits (tested using the MWM) by targeting SYT1 in the ROS-JNK-p53 pathway [25].
1.1. miRNAs Involved in CREB-Dependent Transcription and CREB-Regulated miRNAs
In the invertebrate Aplysia californica, miR-124 is exclusively enriched in sensory neurons, both soma and processes, and regulates serotonin-induced synaptic plasticity via CREB regulation [26]. miR-124 is an abundant, brain-specific miRNA. Reducing miR-124 levels through the injection of an locked nucleic acids (LNA)-probe was found to restore spatial memory and social interaction in adult mice carrying an EPAC-null mutation, whereas AAV-mediated overexpression of miR-124 in the hippocampus impaired long-term potentiation and spatial memory in EPAC+/+ mice by regulating the expression of Zif268 [27], demonstrating the prominent role of miR-124 in the negative regulation of synaptic plasticity and memory formation.
The NAD-dependent deacetylase SIRT1 is essential for normal associative learning in the CFC task, and this function requires crosstalk with the brain-specific miR-134. The transcriptional regulation of miR-134 is repressed by SIRT1, which cooperates with the YY1 DNA-binding element, and miR-134 expression is upregulated in SIRT1-deficient mice. Similar to miR-124, the overexpression of miR-134 negatively regulates fear memory formation and long-term potentiation induction in the rodent hippocampus through translational repression of CREB mRNA; therefore, it affects learning and memory in the CFC paradigm by mediating the CREB-BDNF-dependent signaling pathway [28].
In addition to being a miRNA target, CREB also regulates miRNA transcription. Being co-transcribed from a single locus containing a functional CRE in the promoter, the miR-132/miR-212 cluster can simultaneously be induced by neuronal activity-dependent modulation of CREB [29]. Predictably, miR-132 manipulation was accompanied by changes in memory performance. Lentivirus-mediated interference of miR-132 in the hippocampus or double-knockout miR-132/212 in the forebrain impaired trace fear conditioning, NOR, CFC, and performance in the Barnes maze in mice [29,30], whereas the overexpression of miR-132 in the hippocampus or perirhinal cortex impaired NOR memory in mice [31] and rats [32]. Hansen et al., introduced an inducible miR-132 transgene in the hippocampus of a mouse strain and demonstrated that the expression of relatively low levels of transgenic miR-132 (1.5-fold), which is similar to the physiological induction of miR-132 in spatial memory tasks, could significantly enhance cognitive capacity. In contrast, the overexpression of high levels (3-fold) of miR-132 inhibited learning [33]. This finding indicates that miRNA expression must be maintained within a limited range to ensure normal functioning. Similar to miR-132, we previously found that neuronal activity induces miR-466f-3p through the transcriptional activation of CREB [5]. However, unlike miR-132, which is stress-inducible, miR-466f-3p is only induced in mice with good performance on the MWM task [34]. miR-466f-3p appears to be a positive regulator of neuronal plasticity via the CREB → pCREB → miR-466f-3p → MEF2A axis during spatial learning and memory formation.
1.2. Regulation of Fear Consolidation and Extinction by miRNAs—From Exposure to Inhibitory Learning
Recent examples from the literature further lengthen the list of specific miRNAs thought to be involved in several types of learning-related behaviors and memory formation processes in the mammalian brain, some of which have opposite effects. Several studies have demonstrated a role of miRNAs in amygdala-dependent fear learning. For example, miR-182 was found to be downregulated in the mouse lateral amygdala in vivo after auditory fear conditioning. The overexpression of miR-182 in the lateral amygdala disrupted long-term fear memory [35]. Conversely, miR-182/96/183, which belong to the same miRNA cluster, were induced in the mouse hippocampus during the NOR task training. Mimicking this increase by the overexpression of miR-183/96/182 enhanced object memory, whereas the knockdown of endogenous miR-183/96/182 impaired it. This effect involved the modulation of several neuronal-plasticity-related genes such as HDAC9 [36]. In contrast to miR-182, elevated miR-126a-3p levels contributed to the consolidation of contextual fear memory by modulating its target (EFHD2) in WT mice. Decreasing miR-126a-3p using antagomiR impaired the consolidation of CFC, spatial memory (MWM), and recognition memory (NOR) but not cued fear memory, whereas the overexpression of miR-126a-3p in the dentate gyrus of the hippocampus reduced the Aβ plaque area and neuroinflammation as well as rescued contextual fear memory deficits in the APP/PS1 AD mouse model [37]. The overexpression of miR-135b-3p in the basolateral amygdala not only enhanced remote fear memory in stress-resilient mice but also in the serum of military veterans suffering from post-traumatic stress disorder (PTSD), indicating that miRNAs play roles in fear- and stress-related disorders [38]. In contrast to miR-134 and miR-126a-3p, miR-128b levels in the basolateral amygdala increased only after fear extinction, a learned safety-related fear inhibitory paradigm. The knockdown of miR-128b impaired the formation of fear extinction memory, whereas the forced expression of this miRNA in the mouse infralimbic prefrontal cortex facilitated fear extinction, indicating that miR-128b is specific to this form of inhibitory learning by suppressing genes such as Reelin, Creb1, and Rcs [39]. More specifically, learned safety is a fear inhibitory mechanism that has potential as an experimental model for PTSD and depression. The role of miR-132/-212 in stress-associated, amygdala-dependent learning safety has also been clearly demonstrated [40], suggesting that miRNAs are involved in inhibitory emotional learning and memory. Table 1 and Table 2 summarize studies that have reported the manipulation of miRNA levels in either WT rodents or disease models with changes in cognitive performance.
Table 1. Manipulation of miRNA levels in regulating learning and memory in wild-type rodents.
| miRNA | Change Direction | Method | Fold Change | Location | Behavioral Tasks | Effects in Learning/Memory | Reference |
|---|---|---|---|---|---|---|---|
| miR-34a | Down | miRNA sponge | - | Basolateral amygdala | Auditory fear conditioning | Impaired | [21] |
| Up | Viral overexpression | - | Lateral ventricle | MWM | Enhanced | [22] | |
| Up | Transgenic overepxression | Thousands | Whole brain | T-maze | Impaired | [23] | |
| miR-34c | Up | Viral overexpression | Hippocampus | MWM | Impaired | [41] | |
| miR-92 | Down | miRNA sponge | - | Hippocampus | CFC | Impaired | [42] |
| miR-125b | Up | Injection of mimic | 2 | Hippocampus | CFC | Impaired | [43] |
| miR-126a-3p | Up | Viral overexpression | 1.5~2 | Hippocampus | CFC; MWM; NOR | Enhanced | [37] |
| miR-126a-3p | Down | Virus-mediated interference | 0.4 | Impaired | |||
| miR-128b | Down | Virus-mediated interference | - | Infralimbic prefrontal cortex | Contextual fear extintion | Impaired | [39] |
| Up | Viral overexpression | - | Enhanced | ||||
| miR-132-3p | Up | Transgenic overepxression | 1.5 | Hippocampus | Barnes maze | Enhanced | [33] |
| Up | 3 | NOR; Barnes maze | Impaired | ||||
| Up | Viral overexpression | 2 | Perirhinal cortex | NOR | Impaired | [32] | |
| Down | Virus-mediated interference | Hippocampus | Trace fear conditioning | Impaired | [30] | ||
| Up | Injection of mimic | 2 | basolateral amygdala | Learned safety | Enhanced | [40] | |
| Down | Knock out (KO) | - | Impaired | ||||
| miR-132/212 | Down | Knock out (KO) | excitatory forebrain | NOR; CFC; Barnes maze | Impaired | [29] | |
| miR-134 | Up | Viral overexpression | 6 | Hippocampus | CFC | Impaired | [28] |
| miR-137 | Up | Viral overexpression | - | Hippocampus | CFC; MWM | Impaired | [44] |
| Down | conditional KO | 0.5 | Hippocampus | MWM; Barnes maze | Impaired | [45] | |
| miR-146a | Down | Virus-mediated interference | - | Hippocampus | CFC; NOR; Object location memory test | Impaired | [46] |
| miR-182 | Up | Injection of mimic | 4 | Lateral amygdala | Auditory fear conditioning | Impaired | [35] |
| miR-183/96/182 | Up | Viral overexpression | 4~12 | Hippocampus | NOR | Enhanced | [36] |
| Down | miRNA sponge | 0.5 | Impaired | ||||
| miR-195 | Down | Injection of antagomir | 0.5 | Hippocampus | MWM | Impaired | [47] |
| miR-335-5p | Up | Injection of mimic | - | Hippocampus | MWM | Impaired | [48] |
| miR-466f-3p | Up | Viral overexpression | 1.5 | Hippocampus | MWM; NOR; Barnes maze | Enhanced | [5] |
| Down | miRNA sponge | - | Impaired |
CFC: Contextual fear conditioning; NOR: novel object recognition; MWM: Morris water maze.
Table 2. Manipulation of miRNA levels in regulating learning and memory in transgenic, mutant, or stressed rodents.
| miRNA | Change Direction | Method | Fold Change | Location | Behavioral Tasks | Effects in Learning/Memory | Strains of Disease Model | Reference |
|---|---|---|---|---|---|---|---|---|
| miR-31-5p | Up | Viral overexpression | 3 | Hippocampus | T-maze; NOR; Barnes maze | Enhanced | 3xTg-AD | [49] |
| miR-34c | Down | Injection of inhibitor | - | CFC | Rescued and enhanced | APP/PS1-21 | [24] | |
| Down | Injection of inhibitor | 0.5 | Third ventricle | MWM | Rescued | SAMP8 | [25] | |
| miR-107 | Down | Injection of mimic | 2 | Hippocampus | MWM | Rescued | Aβ ICV injection model | [50] |
| miR-124 | Down | Injection of LNA-probe | 0.4 | Hippocampus | MWM | Rescued | EPAC−/− | [27] |
| Up | Viral overexpression | - | Impaired | EPAC+/+ | ||||
| Up | Viral overexpression | - | Hippocampus | MWM | Impaired | Tg2576 | [51] | |
| Down | Injection of antagomir | - | Rescued | |||||
| miR-124-3p | Up | Viral overexpression | 5~10 | Lateral ventricle | MWM | Rescued | APP/PS1 | [52] |
| miR-126a-3p | Up | Viral overexpression | 1.5~2 | Hippocampus | CFC; MWM; NOR | Enhanced | APP/PS1 | [37] |
| miR-128b | Down | Knock out (KO) | * Cerebral cortex | MWM | Rescued | 3xTg-AD | [53] | |
| miR-132-3p | Up | Viral overexpression | 2.5 | Lateral ventricle | MWM | Rescued | APP/PS1 | [54] |
| Up | Viral overexpression | 1.5 | Hippocampus | MWM | Rescued | Aβ ICV injection model | [55] | |
| miR-132/212 | Up | Injection of mimic | - | Ventricles | Barnes maze | Rescued | 3xTg-AD | [56] |
| miR-134 | Down | Injection of LNA-probe | 0.6 | Rescued | SIRT1 mutant | [28] | ||
| miR-135b | Up | Injection of mimic | 5 | Third ventricle | Y-maze | Rescued | SAMP8 | [57] |
| miR-135b-5p | Up | Viral overexpression | basolateral amygdala | Acute restraint stress and auditory fear conditioning | Enhanced | Stress resilient mice | [38] | |
| Down | Injection of inhibitor | Rescued | Stress susceptible mice | |||||
| miR-139 | Up | Injection of mimic | 2 | Hippocampus | CFC; MWM; NOR | Impaired | SAMP8 | [58] |
| Down | Injection of inhibitor | 0.4 | Rescued | |||||
| Down | Injection of inhibitor | - | Hippocampus | Y-maze; MWM | Rescued | 5xFAD | [59] | |
| miR-181a | Up | Viral overexpression | 4 | Hippocampus | MWM | Rescued | APP/PS1 | [60] |
| miR-188-5p | Up | Viral overexpression | - | Hippocampus | T-maze; CFC | Rescued | 5xFAD | [61] |
| miR-188-3p | Up | Viral overexpression | - | Hippocampus | MWM | Rescued | 5xFAD | [62] |
| miR-195 | Up | Injection of mimic | 2 | Rescued | A chronic brain hypoperfusion model | [47] | ||
| Up | Viral overexpression | - | Hippocampus | MWM | Rescued | APP/PS1 | [63] | |
| Up | Viral overexpression | - | Hippocampus | NOR | Rescued | ApoE4KI +/- 5XFAD | [64] | |
| miR-196a | Up | Viral overexpression | 1.6 | Hippocampus | MWM | Rescued | Aβ ICV injection model | [65] |
| miR-200b/c | Up | Injection of mimic | - | Lateral ventricle | Barnes maze | Rescued | Aβ ICV injection model | [66] |
| miR-206 | Down | Injection of antagomir | - | Cerebral ventricles | CFC; Y-maze | Rescued | Tg2576 | [67] |
| miR-338-5p | Up | Viral overexpression | 2 | Hippocampus | MWM | Rescued | 5xFAD | [68] |
| Up | Viral overexpression | 4~5 | Hippocampus | MWM | Rescued | APP/PS1 | [69] | |
| miR-485-5p | Up | Viral overexpression | 4 | Hippocampus | MWM; CFC | Rescued | APP/PS1 | [70] |
| miR-485-3p | Down | Injection of inhibitor | - | Lateral ventricle | Y-maze | Rescued | 5xFAD | [71] |
CFC: Contextual fear conditioning; NOR: novel object recognition; MWM: Morris water maze. * Only this brain region has been used to performed experiments.
2. Direct Evidence of miRNA Involvement in Cognitive Impairment
2.1. miRNA and AD
There is substantial evidence that miRNAs are involved in the pathophysiology of the age-associated forms of dementia and neurodegeneration. AD is the most common type of dementia in the elderly, and extensive studies on AD have revealed altered miRNA expression profiles. Many miRNAs are severely dysregulated in the brains of AD patients, including miR-34c (upregulated [24]), miR-124-3p (downregulated [72,73]; upregulated [51]), miR-126a-3p (downregulated [72,74]), miR-128b (upregulated [53]), miR-132/-212 (downregulated [74,75,76]), miR-146a (upregulated [72,77]), miR-188-5p (downregulated [61]), miR-195 (downregulated [64]), miR-206 (upregulated [67]), miR-338-5p (downregulated [68]), and miR-485-5p (downregulated [75]) vs. miR-485-3p (upregulated [71]). Figure 1 shows growing evidence that miRNA dysregulation correlates with some major and important aspects of AD pathology that have been proven to cause cognitive impairment in animal models.
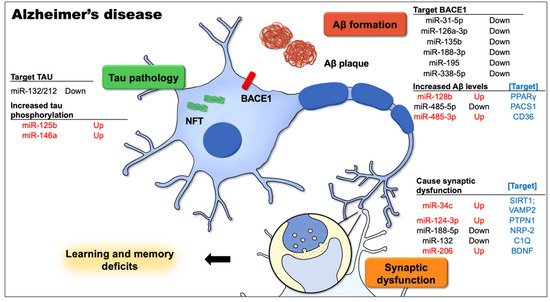
Figure 1. This figure demonstrates the most well-known Alzheimer’s disease (AD)-related cellular and signaling pathways in neurons that contribute to learning and memory deficits, and the dysregulated microRNAs along with the targets involved in the pathophysiological process of AD. The causative factors for AD, including the aggregation of intracellular amyloid-β (Aβ), expression levels and activity of β-site APP cleavage enzyme 1 (BACE1), extracellular amyloid plaques, and neurofibrillary tangles (NFTs) from hyperphosphorylated tau, together with Aβ-mediated synapse elimination and synaptic failure, may lead to cognitive impairment.
2.2. miRNAs Are Involved in Aβ Production and Metabolism
Increasing evidence suggests that miRNAs play a role in regulating Aβ production/metabolism; therefore, Aβ-targeted miRNAs may have therapeutic implications for AD. Aβ peptides are produced from amyloid precursor proteins (APP) after cleavage by the β-site APP cleavage enzyme 1 (BACE1). Several miRNAs, including miR-31-5p, miR-126a-3p, miR-135b, miR-188-3p, miR-195, and miR-338-5p, participate in APP lysis by modulating BACE1 [78,79]. All these miRNAs were found to be downregulated in samples from AD patients or transgenic mice, showing a negative correlation with BACE1, which is highly expressed in the brains of AD patients. The overexpression of hippocampal miR-188-3p reduced BACE1 expression levels and Aβ formation and suppressed neuroinflammation in 5xFAD transgenic mice [62]. The overexpression of miR-126a has been found to be neuroprotective against Aβ42 toxicity, as discussed above [37]. In addition, miR-338-5p, a new miRNA that also targets BACE1, was significantly downregulated in the hippocampus of AD patients and in two animal models, namely 5xFAD and APP/PS1 transgenic mice. The overexpression of miR-338-5p in the hippocampus rescued spatial memory deficits in transgenic mice [68,69]. miR-338-5p is also associated with neuronal differentiation, neurogenesis, and neuronal protective effects through the negative regulation of BCL2L11, which attenuates amyloid plaque deposition, neuroinflammation, and neuronal apoptosis [68,69]. However, there are other miRNAs that increase Aβ levels. For example, miR-128 is involved in the development and progression of AD. In cell culturing, the inhibition of miR-128 attenuated Aβ-mediated cytotoxicity through the inactivation of the NF-κB pathway [80]. The levels of miR-128 and Aβ were significantly increased in the cerebral cortex of 3xTg-AD mice, whereas their target peroxisome proliferator-activated receptor gamma (PPARγ) was downregulated. The knockout of miR-128 attenuated an AD-like performance and alleviated cognitive deficits in 3xTg-AD mice by suppressing amyloid plaque formation, Aβ generation, and neuroinflammation by targeting PPARγ. In addition to animal models, miR-128 has been shown to be upregulated in the brain and plasma samples of AD patients [53,80]. These findings suggest that miR-128 is a useful biomarker for the inflammatory pathophysiology of AD.
2.3. miRNAs Contribute to Abnormal Tau Protein Functions
miRNAs are closely related to the phosphorylation and pathological aggregation of tau proteins. Indeed, as a direct target of miR-132, the knockout of miR-132/212 leads to increased tau expression, phosphorylation, and aggregation. Conversely, the treatment of AD mice with miR-132 partially mimics a restored memory function and tau metabolism [56]. Additionally, the downregulation of miR-132/-212 in the hippocampus and prefrontal cortex of AD patients correlated with neuronal tau hyperphosphorylation, further elucidating the role of miR-132 in tauopathies [75]. Similarly, the expression of miR-132 and miR-212 in neural-derived extracellular vesicles has been shown to be decreased in AD patients [81]. Further, miRNAs can affect tau phosphorylation by regulating the activities of relevant enzymes. The overexpression of miR-125b leads to the upregulation of tau kinases, including p35, CDK5, and p44/42 MAPK (Erk1/2), whereas tau phosphatases (DUSP6, PPP1CA) and the anti-apoptotic factor Bcl-W were downregulated, all of which contribute to tau hyperphosphorylation in primary neurons. Moreover, injection of the miR-125b mimic into the hippocampus of WT mice impaired associative learning in the fear conditioning paradigm and was also accompanied by the downregulation of Bcl-W, DUSP6, and PPP1CA, resulting in increased tau phosphorylation in vivo [43]. The selective knockdown of miR-146a, the most commonly deregulated miRNA in developmental brain disorders, in the hippocampus of adult mice was found to cause severe learning and memory impairments, associated with a reduction in adult hippocampal neurogenesis, indicating for the first time a role for miR-146a in postnatal brain functions [46]. miR-146a was also highly expressed in the brains of AD patients and 5xFAD transgenic mice, and it promoted pathogenesis by modulating the ROCK1/PTEN signaling pathway, resulting in abnormal tau hyperphosphorylation in early NFT. The inhibition of miR-146a expression in the hippocampus resulted in enhanced hippocampal levels of ROCK1, repressed tau hyperphosphorylation, and a partly restored memory function in 5xFAD transgenic mice [59].
2.4. miRNAs Mediate Synaptic Dysfunction
Synapse formation is the basis of neural signal transduction, whereas synaptic plasticity forms the basis of learning and memory. Memory impairment in AD patients results from abnormal synaptic plasticity [16]. Synapses are vulnerable to Aβ-induced neurotoxicity, and the dysregulation of some miRNAs may contribute to defective synaptic elimination and cognition in AD by inducing Aβ-mediated synaptic toxicity.
Kao et al., revealed the role of miR-34c in disrupting dendrites in primary hippocampal neurons [41]. Additionally, miR-34c is upregulated during Aβ accumulation by targeting the VAMP2 gene. As miR-34c blockade upregulated VAMP2 expression levels and rescued Aβ-induced synaptic failure, learning and memory deficits were ameliorated [82]. Another study indicated that miR-124 levels were increased in the temporal cortex and hippocampus of AD patients and in a Tg2576 AD mouse model [51]. Induced levels of miR-124 recapitulated AD-like phenotypes in mice, including memory impairment and deficits in their synaptic transmission and plasticity, by directly regulating the expression of the target gene PTPN1. Thus, maintaining the balance of miR-124/PTPN1 levels by suppressing miR-124 can restore synaptic failure and memory deficits. These findings indicate that the miR-124/PTPN1 pathway is a critical mediator of synaptic dysfunction and memory loss in AD. In contrast, some miRNAs were found to be abnormally reduced in AD models and could have positive effects on neurons. miR-188-5p expression was found to be downregulated in the cerebral cortices and hippocampus of AD patients as well as in the brains of 5xFAD transgenic mice. The replenishment of miR-188-5p rescued Aβ-mediated synapse elimination and synaptic dysfunction, as well as impaired cognitive function by targeting NRP-2 in 5xFAD transgenic mice [61]. One of the targets of miR-132 is C1q, a classical complement cascade protein that mediates synapse elimination in the central nervous system and is highly expressed in AD patients. APP/PS1 transgenic mice transfected with miR-132 showed a significant increase in synaptic protein (PSD95, Synapsin-1, p-Synapsin) expression compared with the non-transfected AD group. These results suggest that miR-132 maintains synaptic plasticity by regulating C1q expression in AD [54].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10081856
This entry is offline, you can click here to edit this entry!