Metabolism plays a fundamental role in both human physiology and pathology, including pancreatic ductal adenocarcinoma (PDAC) and other tumors. Anabolic and catabolic processes do not only have energetic implications but are tightly associated with other cellular activities, such as DNA duplication, redox reactions, and cell homeostasis. PDAC displays a marked metabolic phenotype and the observed reduction in tumor growth induced by calorie restriction with in vivo models supports the crucial role of metabolism in this cancer type. The aggressiveness of PDAC might, therefore, be reduced by interventions on bioenergetic circuits.
- PDAC
- metabolism
- glucose
- amino acids
- lipids
- immune response
1. Introduction
The incidence of pancreatic ductal adenocarcinoma (PDAC) is increasing in recent years and is expected to become the second leading cause of cancer death by 2030 [1]. PDAC is an aggressive tumor often diagnosed at an advanced stage due to the lack of symptoms [2], [3]. PDAC is not always treatable with surgery and in fact only 20% of patients have resectable disease [4], [5]; similarly, chemotherapy treatments are poorly effective. In a cohort of 136 patients, 74.3% experiencing relapse within one year while 25.7% was found to recur in within the first six months of surgery [6]. Next-generation sequencing (NGS) technologies analyzing the mutational profile of PDAC patients recently identified mutations occurring at early or late stages of the disease [7]. KRAS, detected in 90% of cases as mutated and/or amplified [8], is chronologically the first mutated gene in pancreatic intraepithelial neoplasia (PanIN) lesions [9], precancerous stage. The second most commonly mutated gene in PDAC is TP53, predominantly presenting missense mutations [10] associated with a very poor outcome [11]. Another common mutant gene in PDAC is CDKN2A, encoding a cyclin-dependent kinase inhibitor, resulting in loss-of-function alterations; a lower degree of differentiation of PDAC cells was related to a more rapid CDKN2A degradation [12]. SMAD4 mutations occur in late stages of PDAC and seem to be linked to its inactivation in approximately 50% of pancreatic cancer cases, promoting tumor growth and metastasis [13] and representing an increased risk factor for overall survival [14]. The main classification of pancreatic cancer distinguishes between exocrine and neuroendocrine tumors [15], [16]. The vast majority of cases (95%) are exocrine pancreatic cancers including PDAC, which accounts for more than 90%, and acinar cell carcinoma, which makes up 1–2%. Neuroendocrine cancers are rare, comprising less than 5% of all pancreatic cancers [17]. PDAC is often associated with other metabolic comorbidities such as obesity and diabetes, which occur in 15–35% of PDAC patients and are considered risk factors [18], [19], [20]. Algorithms and prediction models were developed to identify high-risk patients among a large number of obese and diabetic patients [21]. Some antidiabetic medications, such as metformin, may decrease the risk of PDAC [22], while others, including insulin, are associated with an increased risk [23]. The fact that PDAC exhibits a marked metabolic phenotype (Figure 1) suggests that the metabolic environment may play a key role [24]. PDAC has high energy requirements, which are met through the rewiring of cell metabolism. Nutrients are therefore consumed to provide energy, ensure biosynthesis, and minimize oxidative stress. PDAC exploits metabolic pathways to sustain rapid cell proliferation [25], thereby depleting major nutrients in the tumor microenvironment and adversely affecting other cell types, in particular immune, acinar, and ductal cells [26]. PDAC cells are surrounded by immune cells, stellate cells, cancer-associated fibroblasts, and extracellular matrix (ECM) [27], [28]. According to their metabolic profile, PDAC cells are divided into three metabolic subtypes: slow proliferating, glycolytic, and lipogenic [29]. Metabolic plasticity also makes a major contribution to cancer heterogeneity [30]. Single-cell RNA sequencing analysis identified distinct types of ductal cells according to their gene expression profiles based on PDAC heterogeneity [31]. A common subtype of ductal cells was found in both healthy and cancerous tissues, while a second subtype showing altered energy distribution was found to reside in PDAC tumors [32]. PDAC is also associated with inflammatory states, which contribute to its progression [33]. Furthermore, inflammation has been linked to the immunometabolic context, since pro-inflammatory stimuli may induce a metabolic switch in hematopoietic cells, increasing aerobic glycolysis similarly to the Warburg effect [34], the well-known shift to aerobic glycolysis (lactate production) in presence of oxygen [35]. Single-cell sequencing suggests that macrophages, T cells, and fibroblasts are highly heterogeneous within the tumor microenvironment [36] and are strongly affected by its metabolic context [37]. In vivo mouse experiments revealed that macrophages exhibited elevated glycolysis, and that macrophage-specific deletion of GLUT-1 reduced tumor burden by increasing natural killer and CD8+ T cell activity and suppressing the inflammatory state [38]. Glutamine antagonists are also reported to induce a change in the antitumor immune response by converting a “cold” tumor microenvironment into a “hot” one, eliciting significant responses to anti-PD1 therapy, a cell surface protein involved in the suppression of the immune system [39]. Taking all these findings together, it seems clear that metabolites play a crucial role homeostasis and tumor progression. The analysis of metabolic turnover in the tumor microenvironment is therefore key to defining the energy phenotype and metabolic landscape of PDAC.
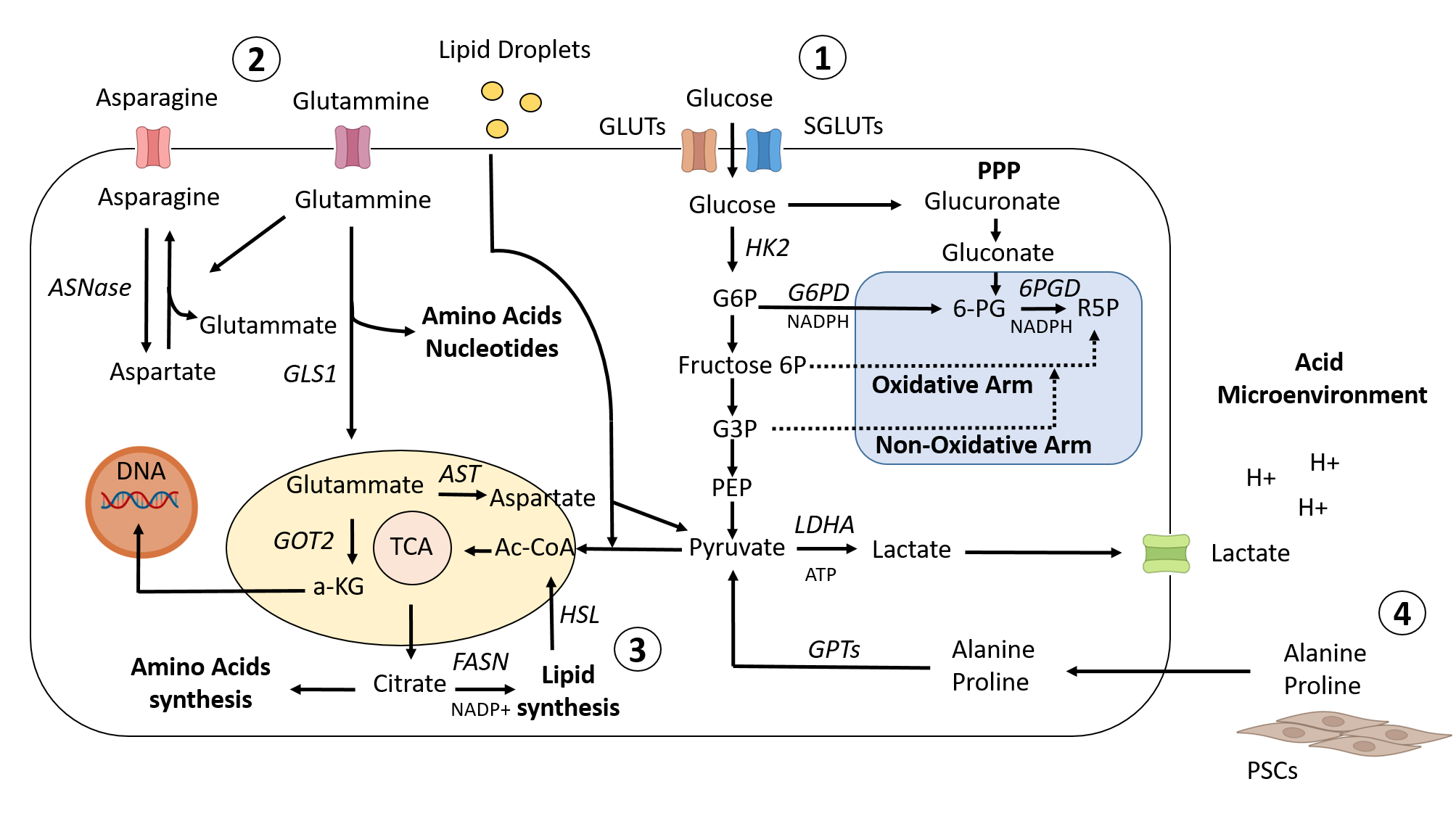
Figure 1. Metabolic landscape in PDAC. 1) To promote glucose uptake in PDAC cells, KRAS and HIF1 upregulate the GLUT family of genes and other genes associated with glycolysis. While a portion of the glycolytic cascade is used to fuel oxidative phosphorylation and the production of ATP, or alternatively to promote lactate, which helps to create an acidic microenvironment, another branch of the process is directed toward the PPP pathway to provide precursors for nucleotide and amino acid biosynthesis. 2) Cellular redox homeostasis and energy generation are both regulated by amino acid metabolism. Glutammine is transformed into glutamate and aspartate, which are then transported to the mitochondria to maintain the redox balance. 3) Citrate is shuttled from the mitochondria into the cytoplasm to stimulate the de novo lipid synthesis pathway, and at the same time, redox processes are balanced by NADPH–NADP+ conversion. This process activates the lipid synthesis pathway. In addition, exogenous lipid intake is boosted to meet the need for nutrients for rapid proliferation. 4) Different metabolites/nutrients, such as Ala and Pro, generated from collagen degradation or PSC secretionand transformed into pyruvate, are supplied to PDAC cells by the tumor microenvironment.
2. Metabolic Reprogramming of the Main Energy Pathways in PDAC
2.1. Activation and Maintenance of Glycoltyc Flux
In PDAC, the expression of glycolytic genes is regulated at both transcriptional and post-transcriptional level via oncogenic KRAS [40]. KRAS signaling plays a crucial role in regulating transcription of both glucose transporters (GLUTs) and key glycolysis genes [41]. Glucose need of the PDAC system seems to be due to the expression of facilitated GLUTS and sodium-glucose transporter (SGLTs); PDAC tumors showed both increased pyruvate carboxylation and glucose oxidation via pyruvate dehydrogenase in vivo [42]. PDAC progression is induced by the activation of mutant KRAS, resulting in an increase in GLUTs, such as GLUT-1, from low- to high-grade dysplasia. Oxygen is related to GLUT-1 expression through hypoxia inducible factor 1 alpha (HIF-1α). In patients with low expression levels of GLUT-1, neoadjuvant chemotherapy such as TS-1 showed better therapeutic response and better prognosis than in those with higher GLUT-1 expression levels [43]. PDAC tumor biology relies on hypoxia and HIF1α signaling to control tumor-promoting stromal programs, which facilitate progression and tumor cell invasivenes [44]. Hypoxia-activated stromal cells contribute to the invasive growth of PDAC cells by releasing soluble proteins, such as MMP10, and enhance the levels of inflammatory and angiogenetic factors including IL1α, TIE family members, and VEGF-A. MMP10, the main stromal protein driving EMT in tumor cells [45], is reported as a stellate cell product [46], [47]. IL1α was shown to be released by both stromal cells and PDAC cells, thus promoting tumor growth [48] in an autocrine manner [49], and stimulated the fibrotic component [50]. TIE1 upregulation and increased TIE2 transcription in hypoxic stellate cells are crucial for the remodeling and maturation of tumor vasculature [51], forming a complex with angiopoietins and sustaining TIE2 signaling in contacting cells [52]. Altered levels of VEGF-A found in PDAC indicate an imbalance in normal angiogenetic processes [53]. In particular, the high extracellular matrix component associated with vasculature collapse resulted in an increased hypoxic environment, partly explaining the low efficacy of antiangiogenic drugs in this cancer [54] and the inefficient delivery of chemotherapeutic agents [55]. Thus emphasizing a recently described stroma-targeting therapy that aims to reduce the stromal component to improve target achievement [56]. According to the transcriptomic profiles of PDAC patients, ubiquitin specific peptidase 25 (USP25) depletion was linked to decreased levels of HIF-1α, GLUT-1, and glycolysis signaling. This suggests that USP25 complex deubiquitinates and stabilizes the HIF-1 α transcription factor from a mechanistic point of view [57]. SGLTs also play a functional role in glucose uptake, since the selective inhibition of SGLT2 in mouse models of pancreatic cancers led to a decrease in glucose uptake [58]. Furthermore, the hypoxic environment is essential for maximizing energy yield and biomass production, which are ensured by the lack of oxygen, which promotes conversion of pyruvate into lactate by Lactate Dehydrogenase A [59]. In such way, ATP is generated and an increased amount of lactic acid is released outside of the cell, acidifying the microenvironment and in turn facilitating PDAC progression [60]. Monocarboxylate transporters (MCTs), which transport lactate, are abundantly expressed in PDAC [61]. MCT1 and MCT4 regulate lactate efflux through KRAS-dependent signaling, releasing intracellular accumulated lactate and maintaining intracellular pH [62]. This process facilitates the oxidation of nicotinamide adenine dinucleotide (NADH) to NAD+, a cofactor for oxidizing glyceraldehyde 3-phosphate and driving glycolysis [63]. The glycolytic shift meets the energy demands required for tumor growth, as well as supplying the building blocks for biochemical reactions and intermediates [64]. MUC1 and MUC13 transporters also stabilize transcription of HIF-1α during hypoxia conditions and induce the expression of glycolytic genes [65] associated with poor survival rates in PDAC patients [57]. In contrast, CD147 works as a chaperone for the membrane localization of MCT1 and MCT4, both expressed in PDAC cells [66]. Whether the interaction between CD147 and MCT is related to PDAC progression has not yet been determined [67]; however, depletion of MCT4 reduces cell viability, whereas depletion of CD147 affects tumor growth in xenograft models [62], [68]. As regards the glycolytic flux of anabolic pathways in PDAC, the pentose phosphate pathway (PPP) is a branch of glycolysis that directs glucose flux to oxidation and regulates NADP and nucleic acid synthesis, which ensure fatty acid (FA) production and cell survival under stress conditions [69]. According to a metabolomic analysis of PDAC, the adaptation to acidosis status increases glucose and decreases glycolysis, driving a shift to PPP [70], [60]. PPP occurs in two different ways: oxidatively and non-oxidatively. The oxidative arm transforms glucose 6-phosphate into ribulose-5-phosphate and CO2, which are essential for maintaining redox equilibrium under stress conditions [71]. The non-oxidative branch produces glycolytic intermediates, resulting in the production of sugar phosphate, an important precursor for amino acid synthesis, ribose-5-phosphate which is needed for nucleic acid synthesis [72]. Furthermore, oncogenic KRAS selectively activates non-oxidative PPP, possibly via the induction of genes involved in the non-oxidative arm, such as ribulose-5-phosphate isomerase (RPIA) [73]. Low expression levels of RPIA deficits result in reduced KRAS-driven signaling in PDAC cells, indicating the importance of non-oxidative PPP in metabolic function [40]. Growing evidence suggests that non-oxidative PPP contributes to gemcitabine resistance in PDAC and that reduced expression of transketolase is associated with higher gemcitabine sensitivity in PDAC patients, strengthening the therapeutic potential of targeting non-oxidative PPP [65]. Post-transcriptional processes, such as those modulated by microRNAs, are also thought to play an important role in PDAC progression [74]. microRNAs are linked to the regulation of glycolysis in PDAC; the tumor suppressor miR-124 regulates MCT1 [75], resulting in increased intracellular pH that reduced acidic environment and decreased PANC-1 cell proliferation. miR-135 was found significantly overexpressed in PDAC patient samples compared to normal tissue, and notably associated to a metabolic alteration. It has been observed miR-135 accumulation during glutamine deprivation, promoted by mutant TP53. Specifically, miR-135 targets phosphofructokinase-1 inhibiting aerobic glycolysis and promoting TCA cycle [74]. Some studies have already been conducted on the potential role of mirna as biomarkers of PDAC. miRNA-483-3p and miRNA-21 were found to be significantly higher from blood plasma in PDAC compared to healthy controls, and related to to advanced stage disease [76], [77]. Further functional studies on miR-124 may lead to new therapeutic strategies for PDAC [75].
2.2. Amino Acids as an External Energy Resource
The PDAC phenotype is also triggered by the rewiring of amino acids, contributing to the metabolic profile of PDAC by regulating cell proliferation, invasion, and redox homeostasis [78]. In cellular hemostasis, glutamine is a multifunctional amino acid that serves as a key energy source [79]. The biological activities of glutamine rangefrom providing energy to stabilizing reducing agents, contributing to the biosynthesis of purines and pyrimidines, and its involvement in PDAC has been recognized [80], [81]. PDAC cells can compensate for the increased metabolic demand either by increasing glutamine production or by increasing glutamine uptake from the environment, thus reducing glutamine levels in blood serum despite the abundance of fibrotic cells in the pancreas [82]. Glutamate-Ammonia Ligase (GLUL), the enzyme responsible for de novo production of glutamine, was found elevated in PDAC [83]. Although the cause of this increase is not completely clear, CRISPR/Cas9 ablation of GLUL in PDAC mouse models reduced tumor growth [83]. Metabolic niches also contribute significantly to cancer development and progression. Autophagy plays a pivotal role in supporting the growth of PDAC through fibroblasts [84]. Autophagy allows fibroblasts to break down misfolded proteins and ECM, releasing large quantities of amino acids into the microenvironment [85]. In addition, circulating macromolecules enter PDAC cells using using the Na+-dependent glutamine transporter SLC1A5, in the case of glutammine, or via macropinocytosis/micropinocytosis, for proteins, a mechanism linked to the growth of cancer cells expressing oncogenic KRAS [86], [87], [88], [89]. Micropinocytosis inhibitors were found to interfere with this ability in MIAPaCa2 cells, a PDAC model [90]. Glutamine intake is converted into glutamate to feed a complex network of enzymes and intermediates. PDAC utilizes glutamate to activate the tricarboxylic acid (TCA) cycle and electron transport chain after its conversion into alpha-ketoglutarate (αKG) in mitochondria; notably, αKG acts as an epigenetic factor [91]. αKG may also function in a TCA-independent manner by acting as a cofactor for dioxygenases, controlling gene expression, DNA methylation, and DNA damage reactivity [92]. Similarly to glutamine, alanine is also required for metabolic homeostasis in PDAC and is derived from pancreatic stellate cells (PSCs) [93]. Several studies have investigated the unidirectional channeling of alanine between PSCs and PDAC [93], [94]. SLC38A2 activity facilitates alanine uptake, although other transporters have been identified, including SLC1A4 [93]. PDAC cells also express the mitochondrial isoform of glutamic-pyruvic transaminase ALT2 for de novo synthesis and alanine utilization. The ratio between aspartate transaminase AST and alanine aminotransferase ALT was used to predict poor prognosis and response to gemcitabine/nab-paclitaxel treatment in PDAC patients [95]. In co- injection xenograft models, the beneficial support provided by stellate cells was disrupted by targeting SLC38A2, causing significant tumor regression in PDAC and affecting cytosolic alanine internalization and concentration [93]. PDAC can also use proline as a fuel source, and this energy comes from collagen that is largely found in the ECM [96]. Proline degradation by the mitochondrial enzyme PRODH1 plays as an active factor in PDAC cell proliferation both in vitro and in vivo [96], indicating that ECM is an important nutrient reservoir for cancer cell metabolic flexibility. Some context-specific metabolic mechanisms have also been described for PDAC, such as the TP53-mediated overexpression of SLC1A3, an Na+/K+/H+-dependent aspartate/glutamate transporter, which enables the aspartate metabolism to maintain cancer cell survival and tumor growth under conditions of glutamine starvation [97]. By perturbing glutamine metabolism, redox homeostasis proteins are deregulated, leading to reactive oxygen species ROS accumulation, which then leads to a cellular redox imbalance facilitating PDAC cell apoptosis [98]. Pharmacological and genetic targeting of nicotinamide phosphoribosyltransferase (Nampt), a key redox enzyme, inhibited cell growth and survival of PDAC cells in vitro and in vivo [99]. Other findings link amino acids with cell fate. KRAS-driven PDAC mouse models was less responsive to depletion of serine and glycine [100]. Cysteine depletion induced ferroptosis in KRAS/TP53 mutant pancreatic tumors in mice, and the disruption of amino acid pathways was able to enhance gemcitabine chemosensitivity in drug-resistant PDAC [101], [98]. Ferroptotic damage can result in the of release damage-associated molecular pattern molecules, which can lead to inflammation [102].
2.3. Fatty Acids Contribute to PDAC Progression
Epidemiological studies correlated PDAC with dyslipidemia [103], showing an altered biosynthesis of cholesterol and other lipids in murine PDAC cells [104], [105], [106], [107]. Lipogenic enzymes are frequently overexpressed in PDAC, supporting their potential contribution to tumor growth [108]. Alanine from PSCs can be taken up by PDAC cells and used for FA biosynthesis. Serum FA synthase (FASN) levels are in fact generally higher in PDAC patients [109] as a result of SREBP1 activity [110] and are associated with lower survival than in patients with low FASN expression and with poor response to gemcitabine [111], [112]. Once again, driver mutations in KRAS and loss-of-function in TP53 reprogram metabolism, accelerating cholesterol biosynthesis and uptake [40], mediating metabolic plasticity via SREBP1-dependent regulation of transforming growth factor-β expression involved in PDAC differentiation. Oncogenic KRAS regulates hormone-sensitive lipase (HSL) to control metabolism by regulating lipid storage and utilization (specifically through suppression of HSL expression), leading to lipid droplet (LD) accumulation and priming tumor cells for invasion [113]. Perilipins constitute the major proteins resident on LD surface controlling intracellular lipid homeostasis [114], [115]. Perilipin 2 (PLIN2) was found overexpressed in a cohort of 181 PDAC patients [116] and was associated with poor MFS, DFS, and OS rates as well as with poor prognosis. Further investigations using an in vivo mouse model showed that exposure of pancreatic β cells to fatty acids stimulated PLIN2 expression, impacting on cellular stress, whereas its ablation prevented fatty acid-induced TG accumulation [115], mitigating stress and leading to a significant improvement of hyperglycemia [117]. Notably, PLIN2 is expressed in other cell types such as monocytes and macrophages [118], where its expression was positively correlated with LGALS9 in PDAC; this protein converts polarized macrophages into an M2 phenotype, leading to the inhibited secretion of T cell cytokines [119]. These findings suggest that PLIN2 might participate in immunomodulatory effects by regulating tumor-associated macrophages in the tumor microenvironment [120]. A high fat diet was able to ameliorate mutated Kras activity, increasing fibrosis and enhancing PDAC progression in a mouse model [121], and a recent study with in vivo mouse model showed a causal and positive corrrelation between obesity and early PDAC progression, identifying altered beta cell expression of cholecystokinin (Cck) in response to obesity defining islet Cck as a promoter in oncogenic Kras-driven PDAC [122]. LDs are recognized as important regulators in cancer; these dynamic intracellular organelles are used for cellular storage of lipids such as triacylglycerol and cholesterol ester [123]. Lipids can thus be catabolized by lipolysis via lipases to liberate free FAs [124], causing increased FA oxidation and oxidative metabolism, which drives tumor cell invasion. Low-density lipoprotein receptor (LDL-R) is highly expressed in PDAC and is associated with increased PDAC recurrence [125]. LDL-R increases cholesterol uptake, while its inhibition reduces proliferation, affecting ERK1/2 survival pathway, and sensitizes PDAC cells to chemotherapeutic drugs, favoring tumor regression [125]. Interestingly, mutated KRAS is able to control the sequestration of extracellular unsaturated FAs [126]. ACSL3 activity, a protein coding gene for a member of Acyl-CoA synthetase long chain family, has been linked to KRAS-mutated tumors [127] and associated with the retention of extracellular unsaturated FAs by converting them into esters that remain confined in PDAC cells [128], [129]. Serum lipid depletion or ACSL3 inhibition decreased tumor cell proliferation, provoking a rebound effect due to lipid restriction that was balanced by increased autophagic flux, in both in vitro and in vivo models [130]. Notably, combining lipid depletion with autophagy inhibitors induced the most potent effect, with arrest of PDAC proliferation and increased apoptosis [130]. Recently, metabolomic profiles clarified key aspects of the metabolic signature of pancreatic cancer stem cells (PCSCs) originating from PDAC cells, revealing a fundamental role for pyruvate-malate cycle and lipid metabolism in their survival [131]. While lipidomic analysis suggested a strong induction of long chain FAs and accumulation of LDs mediated by ELOVL5, a fatty acid elongase, other data highlighted cardiolipin acyl-chain composition as pivotal in PCSCs [132]. Changes in cardiolipin composition have an impact on enzymes involved in the respiratory process and integrity of the inner membrane [133], [134], indicating that cardiolipin plays a critical role in oxidative phosphorylation. A comprehensive investigation on serum lipids of 830 PDAC samples by mass spectrometric determination revealed statistically significant differences between PDAC patients and healthy controls [135]. While a lysophosphatidylcholine LPC 18:2 was positively correlated with survival, Cer 36:1, Cer 38:1, Cer 42:2, PC 32:0, PC O-38:5, and SM 42:2 were inversely correlated, suggesting their potential role as prognostic biomarkers. Other data in PDAC tissues by MALDI-MSI analyses indicated that LPC (16:0, 18:1), as reported for other LPCs [136], [137], and DAG 36:2 were decreased, while PC 32:0, SM d36:1, and SM d42:3 were increased [138]. Glycerophospholipid and sphingolipid metabolism pathways were also found dysregulated in PDAC [138]. About lipid saturation degree, polyunsaturated phosphatidylcholines were reduced in serum of PDAC [139] it is tempting to speculate that this altered profile might reflect apoptotic resistance in PDAC, given that polyunsaturated FAs, via peroxidation, act as substrates for ferroptosis in cell membranes [140], [141].
3. Immune Cells and Metabolic Response in PDAC Microenvironment
Immune cell functionality and metabolism are closely linked and are able to influence each other [142]. In recent years, several studies provided compelling evidence that changes in cellular metabolism affect immune cell function (Figure 2), which in turn impacts cell metabolism [143], [142] due to competition for nutrients. The high energy demands of tumor cells cause nutrient depletion, resulting in decreased rates of glycolysis in tumor-infiltrating lymphocytes [144], [145]. The imbalance in metabolic profile and chronic inflammation can trigger autoreactivity and ultimately disrupt protective immunity. Several immunosuppressive cytokines were found to cause tumor development by impairing cytotoxic and helper T cells [146]. The acid environment created by lactic acid levels inhibited cytotoxic T cell function, promoting tumor growth [147]. In addition, immunohistochemistry analysis detected an increase in CD8+ and CD4+ T cells, leading to a better outcome in PDAC patients [148], [149]. In PDAC, CD8+ T cell activity is altered due to degradation via MHC-1, member of histocompatibility complex, [150], while inhibition of autophagy restores surface levels of MHC-1 enhancing anti-tumor T cell response improving [151] clinical outcomes, increasing the survival rate for PDAC patients [152]; the effect observed in CD4+ T cells depends on their subtype differentiation [153]. T-helper 1 cells contributed to a positive clinical outcome via IFN-γ and TNF-α production, promoting anticancer activities through cytotoxic T cell response [154], [155]. Interestingly, insulin receptors are expressed on activated CD4+ T cells and can contribute to reshaping the adaptive immune system by regulating T cell metabolism [156]. Induced knockdown of insulin receptors led to a reduced glucose metabolism and cytokine production in T cells [157]. Glutamine may also act as a regulator of effector and regulatory T cell (Treg) balance in the PDAC microenvironment, reducing Th17 and Th1 cells and promoting the development of Tregs [158]. The production of αKG by glutamate dehydrogenase promotes cancer growth and interferes with immune response, acting as an anaplerotic intermediate in the TCA cycle and providing nitrogen for non-essential amino acid synthesis [81]. T cells need arginine and tryptophan for activation to generate memory T cells by switching from glycolysis to oxidative phosphorylation, thus promoting tumor arrest [159]. PDAC is thought to be fueled by a feed forward mechanism in which amino acid intermediates support cancer growth by depleting arginine and tryptophan, inhibiting T cell proliferation and promoting Treg differentiation [160]. Indeed, Treg can inhibit immune responses mediated by T cells, as described in a study involving a total of 100 patients with PDAC [161]. The authors evaluated the prevalence of Treg in peripheral blood mononuclear cells from patients in relation to their clinical outcomes and showed that the percentage of Treg in the patients with PDAC was significantly lower than in healthy volunteers. Additionally, numbers of mast cells from 103 patients with PDAC and 10 patients with normal pancreas have been investigated about their distribution [162]. Results showed a zone-specific distribution of mast cells in PDAC highlighting the importance of invasive front in the prognosis of patients with PDAC after curative resection. Dendritic cells are an integral part of the PDAC tumor microenvironment characterized by reduced number compared to healthy condition, which impacts antigen presentation and contributes to immune tolerance [163]. Macrophages have also been linked to the immunometabolic context in PDAC. In response to the environment conditioned by PDAC, macrophages switch from the pro-inflammatory M1 to the anti-inflammatory M2 phenotype [164], [165]. M1 macrophages showed an enhanced glycolytic and lipolytic activity [166] promoted by fructose-2,6-bisphosphatase enzyme [167]. Furthermore, when PPP is inhibited macrophages switch toward an anti-inflammatory state, increasing TCA cycle and FA oxiydation [168], [169], [170].
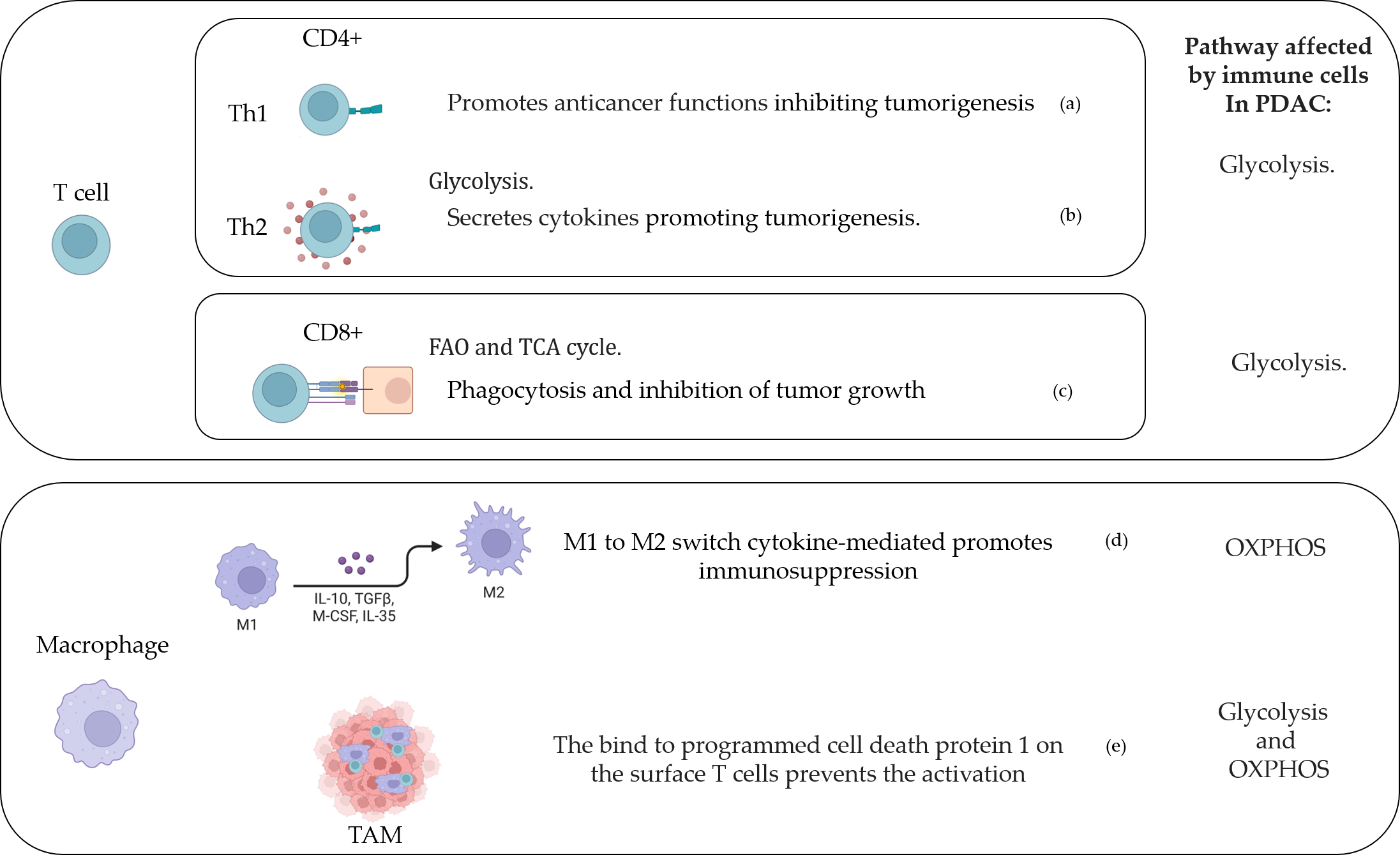
Figure 2. T cell and macrophage activity in the PDAC-conditioned environment. (a) [154], [169]; (b) [171]; (c) [148], [172]; (d) [164]; (e) [173]. Created with BioRender.com.
4. Clinical perspectives
Over the last decade, it has become clear that rapidly proliferating systems such as cancer use metabolism to facilitate cell survival and maintain growth [174]. Metabolic plasticity contributes to PDAC heterogeneity, and although not all metabolic dependencies of pancreatic tumors have been revealed, some distinct phenotypes, such as the glycolytic and lipogenic subtypes, have been identified. As a consequence, potentially innovative strategies to treat patients with PDAC are based on glycolytic and glutamine inhibitors, which have shown efficacy for the glycolytic subtype, while the lipogenic phenotype is more sensitive to inhibitors of lipid biosynthesis. Currently, PDAC trials are mainly focused on immunotherapy and chemotherapy [174], and there are still few trials investigating cell metabolism. Here the researchers reported the clinical trials focused on metabolic evaluation in PDAC (Table 1), such as NCT05132244 that will investigate the feasibility to monitor and manage glucose levels in people with PDAC and the related impact. NCT04245644 aims to understand if regular use of statins and metformin can increase rate of disease-free survival and overall survival in PDAC participants, before diagnosis, after surgery and in neo-adjuvant treatment setting, and possible use as chemoprevention. While a large randomized study of 528 participants (NCT03504423) is evaluating the effect of CPI-613 (devimistat), a pyruvate dehydrogenase inhibitor, to determine its efficacy and safety in patients with metastatic PDAC. The aim of another study (NCT02201381) is to assess the effectiveness of a regimen of metabolic treatments for patients in order to determine the relationship between degree of response and changes in biochemical markers. In a cohort of 207 participants, metformin, atorvastatin, doxycycline, and mebendazole will be administered to evaluate the effectiveness of heterogeneous classes of drugs. Pharmacologically, metformin improves insulin sensitivity and the oxidative disposal of glucose and lactate. NCT04862260 will investigate the effect on cholesterol disruption also in metastatic PDAC patients (Figure 3). Statins lower cholesterol by inhibiting HMG-CoA reductase, the rate-limiting enzyme of the metabolic pathway producing cholesterol and other isoprenoids, thereby blocking lipid flow. Glycolytic and oxidative metabolisms can both be altered by doxycycline, while the carbohydrate metabolism can be affected by mebendazole. Devimistat already showed antitumor activity in xenograft mouse models of human colorectal cancer, enhancing therapeutic efficacy and preventing irinotecan-triggered p53 stabilization, making it a promising candidate to support antineoplastic therapy [175]. Other drugs for non-oncological use have shown off-label efficacy in PDAC, modulating proliferative arrest [176]. When drug targets such as GLUT-1 were knocked out, a strong growth-inhibiting effect on PDAC biomass was observed, resulting in a no-growth phenotype [177].
| Identifier ID | Study Title | Conditions | Interventions |
|---|---|---|---|
| NCT05296421 | Investigating Targetable Metabolic Pathways Sustaining Pancreatic Cancer | Primary | Procedure: Biopsy, Therapeutic Conventional Surgery Other: Uniformly-labeled [13C] glucose |
| NCT04565327 | Hyperpolarized 13C Pyruvate MRI for Treatment Response Assessment in Patients With Locally Advanced or Metastatic Pancreatic Cancer | Primary | Drug: Hyperpolarized Carbon C 13 Pyruvate, Procedure: Magnetic Resonance Imaging (MRI) |
| NCT04862260 | Cholesterol Disruption in Combination With FOLFIRINOX in Patients With Metastatic Pancreatic Adenocarcinoma | Primary and Metastatic | Drug: Cholesterol metabolism disruption |
| NCT02978547 | The Effects of Neoadjuvant Metformin on Tumor Cell Proliferation and Tumor Progression in Pancreatic Ductal Adenocarcinoma | Primary | Drug: Metformin Hydrochloride 500 Mg Tablet |
| NCT05254171 | Study of Nab-Paclitaxel and Gemcitabine With or Without SBP-101 in Pancreatic Cancer | Primary | Drug: SBP-101, Nab-paclitaxel, Gemcitabine and Placebo |
| NCT03450018 | A Study of SLC-0111 and Gemcitabine for Metastatic Pancreatic Ductal Cancer in Subjects Positive for CAIX | Metastatic | Drug: SLC-0111, Gemcitabine Injection |
| NCT05132244 | Monitoring and Managing Glucose Levels in People With Pancreatic Cancer | Primary | Procedure: Endocrinologist-directed target blood glucose level 4–10 mmol/L using data from a continuous glucose monitor (CGM). Other: Standard Care |
| NCT04915417 | Neoadjuvant Stereotactic Ablative Radiotherapy for Pancreatic Ductal Adenocarcinoma | Primary | Radiation: Stereotactic Ablative Body Radiotherapy (SABR) |
| NCT04662879 | Early Detection Initiative for Pancreatic Cancer | Primary | Other: Enriching New-onset Diabetes for Pancreatic Cancer (ENDPAC) score. Other: Abdominal imaging |
| NCT03525392 | Study to Evaluate the Safety and Activity (Including Distribution) of 177Lu-3BP-227 in Subjects With Solid Tumors Expressing Neurotensin Receptor Type 1. | Primary | Drug: 177Lu-3BP-227 (also called 177Lu-IPN01087) |
| NCT03410030 | Trial of Ascorbic Acid (AA) + Nanoparticle Paclitaxel Protein Bound + Cisplatin + Gemcitabine (AA NABPLAGEM) | Primary | Drug: Ascorbic Acid, Paclitaxel protein-bound, Cisplatin, Gemcitabine |
| NCT04245644 | Efficacy of Chemopreventive Agents on Disease-free and Overall Survival in Patients With Pancreatic Ductal Adenocarcinoma: The CAOS Study (CAOS) | Primary | Behavioral: use of targeted drugs such as aspirin, B-Blockers, Metformin, ACE-inhibitors, Statins |
| NCT03374852 | CPI-613 in Combination With Modified FOLFIRINOX in Patients With Locally Advanced Pancreatic Cancer | Primary | Drug: CPI-613 Drug: mFOLFIRNOX |
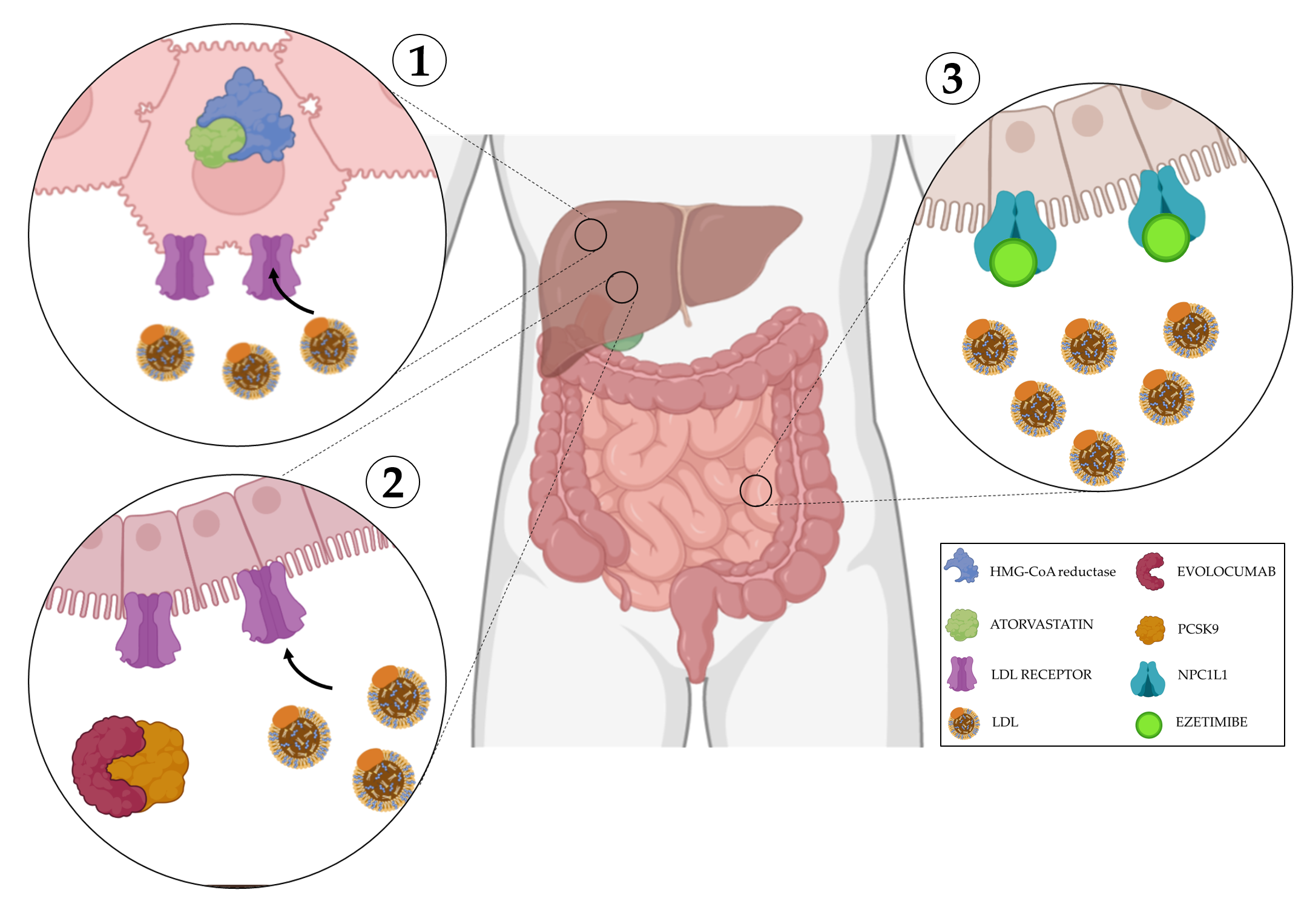
Figure 3. Mechanisms of action of cholesterol disruption. 1) Atorvastatin is a competitive inhibitor of HMG-CoA reductase, its inhibitiondecreases de novo cholesterol synthesis and increase expression of LDL receptors, removing LDL from the blood. 2) Evolocumab blocks PCSK9, a protein responsible for the breakdown of LDL receptors, this allows their overexpression facilitating LDL uptake from the blood. 3) Ezetimibe selectively inhibits the instestinal absorption of LDL. Created with BioRender.com.
5. Conclusions
Here, the researchers discuss the metabolic profile of PDAC and its implications in terms of clinical outcomes. Blocking anabolic and catabolic processes is able to reduce PDAC progression, validating the plausible hypothesis that PDAC relies on metabolic reprogramming. Although the findings presented here identify metabolic processes as a potential target for this tumor, the translation of this approach to the clinic is slow, and clinical trials investigating metabolic reprogramming in PDAC are still few and far between. Exploring the metabolic landscape could lead to the development of innovative therapeutic strategies, increasing the chances of successful treatment and survival.
Abbreviations:
alpha-ketoglutarate (αKG); cholecystokinin (Cck); extracellular matrix (ECM); FA synthase (FASN); fatty acid (FA); glutamate-ammonia ligase (GLUL); glucose transporters (GLUTs); hormone-sensitive lipase (HSL); hypoxia inducible factor 1 alpha (HIF-1α); lipid droplet (LD); Low-density lipoprotein receptor (LDL-R); Monocarboxylate transporters (MCTs); Next-generation sequencing (NGS); nicotinamide adenine dinucleotide (NADH); nicotinamide phosphoribosyltransferase (Nampt); pancreatic ductal adenocarcinoma (PDAC); ancreatic cancer stem cells (PCSCs); pancreatic intraepithelial neoplasia (PanIN); pancreatic stellate cells (PSCs); pentose phosphate pathway (PPP); ribulose-5-phosphate isomerase (RPIA); sodium-glucose transporter (SGLTs); tricarboxylic acid cycle (TCA); ubiquitin specific peptidase 25 (USP25)
This entry is adapted from the peer-reviewed paper 10.3390/cancers14153799
References
- Andrew McGuigan; Paul Kelly; Richard Turkington; Claire Jones; Helen G Coleman; R Stephen McCain; Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World Journal of Gastroenterology 2018, 24, 4846-4861, 10.3748/wjg.v24.i43.4846.
- Prashanth Rawla; Tagore Sunkara; Vinaya Gaduputi; Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World Journal of Oncology 2019, 10, 10-27, 10.14740/wjon1166.
- Terumi Kamisawa; Laura D Wood; Takao Itoi; Kyoichi Takaori; Pancreatic cancer. The Lancet 2016, 388, 73-85, 10.1016/s0140-6736(16)00141-0.
- Anastasios Stathis; Malcolm J. Moore; Advanced pancreatic carcinoma: current treatment and future challenges. Nature Reviews Clinical Oncology 2010, 7, 163-172, 10.1038/nrclinonc.2009.236.
- Jayme B. Stokes; Norris J. Nolan; Edward B. Stelow; Dustin M. Walters; Geoffrey R. Weiss; Eduard E. De Lange; Tyvin A. Rich; Reid B. Adams; Todd W. Bauer; Preoperative Capecitabine and Concurrent Radiation for Borderline Resectable Pancreatic Cancer. Annals of Surgical Oncology 2011, 18, 619-627, 10.1245/s10434-010-1456-7.
- Toshitaka Sugawara; Daisuke Ban; Jo Nishino; Shuichi Watanabe; Aya Maekawa; Yoshiya Ishikawa; Keiichi Akahoshi; Kosuke Ogawa; Hiroaki Ono; Atsushi Kudo; et al. Prediction of early recurrence of pancreatic ductal adenocarcinoma after resection. PLOS ONE 2021, 16, e0249885, 10.1371/journal.pone.0249885.
- Gong-Qing Shen; Essa M. Aleassa; R. Matthew Walsh; Gareth Morris-Stiff; Next-Generation Sequencing in Pancreatic Cancer. Pancreas 2019, 48, 739-748, 10.1097/mpa.0000000000001324.
- Sebastian Mueller; Thomas Engleitner; Roman Maresch; Magdalena Zukowska; Sebastian Lange; Thorsten Kaltenbacher; Björn Konukiewitz; Rupert Öllinger; Maximilian Zwiebel; Alex Strong; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62-68, 10.1038/nature25459.
- Andrew Waters; Channing J. Der; KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harbor Perspectives in Medicine 2017, 8, a031435, 10.1101/cshperspect.a031435.
- A Scarpa; P Capelli; K Mukai; G Zamboni; T Oda; C Iacono; S Hirohashi; Pancreatic adenocarcinomas frequently show p53 gene mutations.. The American Journal of Pathology 1993, 142, 1534-43, .
- Martino Maddalena; Giuseppe Mallel; Nishanth Belugali Nataraj; Michal Shreberk-Shaked; Ori Hassin; Saptaparna Mukherjee; Sharathchandra Arandkar; Ron Rotkopf; Abby Kapsack; Giuseppina Lambiase; et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proceedings of the National Academy of Sciences 2021, 118, 4, 10.1073/pnas.2025631118.
- Nilabja Sikdar; Gourab Saha; Ashmita Dutta; Shibajyoti Ghosh; Shailesh V. Shrikhande; Sudeep Banerjee; Shailesh V. Shreekhande; Genetic Alterations of Periampullary and Pancreatic Ductal Adenocarcinoma: An Overview. Current Genomics 2018, 19, 444-463, 10.2174/1389202919666180221160753.
- Nabeel Bardeesy; Kuang-Hung Cheng; Justin H. Berger; Gerald C. Chu; Jessica Pahler; Peter Olson; Aram F. Hezel; James Horner; Gregory Y. Lauwers; Douglas Hanahan; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes & Development 2006, 20, 3130-3146, 10.1101/gad.1478706.
- Christopher H. Crane; Gauri R. Varadhachary; John S. Yordy; Gregg A. Staerkel; Milind M. Javle; Howard Safran; Waqar Haque; Bridgett D. Hobbs; Sunil Krishnan; Jason B. Fleming; et al. Phase II Trial of Cetuximab, Gemcitabine, and Oxaliplatin Followed by Chemoradiation With Cetuximab for Locally Advanced (T4) Pancreatic Adenocarcinoma: Correlation of Smad4(Dpc4) Immunostaining With Pattern of Disease Progression. Journal of Clinical Oncology 2011, 29, 3037-3043, 10.1200/jco.2010.33.8038.
- Milena Ilic; Irena Ilic; Epidemiology of pancreatic cancer. World Journal of Gastroenterology 2016, 22, 9694-9705, 10.3748/wjg.v22.i44.9694.
- Daniel Ansari; Bobby Tingstedt; Bodil Andersson; Fredrik Holmquist; Christian Sturesson; Caroline Williamsson; Agata Sasor; David Borg; Monika Bauden; Roland Andersson; et al. Pancreatic cancer: yesterday, today and tomorrow. Future Oncology 2016, 12, 1929-1946, 10.2217/fon-2016-0010.
- Eric A. Collisson; Peter Bailey; David K. Chang; Andrew V. Biankin; Molecular subtypes of pancreatic cancer. Nature Reviews Gastroenterology & Hepatology 2019, 16, 207-220, 10.1038/s41575-019-0109-y.
- Rachel R Huxley; Alireza Ansari-Moghaddam; A Berrington De González; Federica Barzi; Martin J Woodward; Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. British Journal of Cancer 2005, 92, 2076-2083, 10.1038/sj.bjc.6602619.
- C. Bosetti; V. Rosato; D. Li; D. Silverman; G. M. Petersen; P. M. Bracci; R. E. Neale; J. Muscat; K. Anderson; S. Gallinger; et al. Diabetes, antidiabetic medications, and pancreatic cancer risk: an analysis from the International Pancreatic Cancer Case-Control Consortium. Annals of Oncology 2014, 25, 2065-2072, 10.1093/annonc/mdu276.
- Sara H. Olson; Youming Xu; Keri Herzog; Amethyst Saldia; Ersilia M. DeFilippis; Peter Li; Peter J. Allen; Eileen M. O'Reilly; Robert C. Kurtz; Weight Loss, Diabetes, Fatigue, and Depression Preceding Pancreatic Cancer. Pancreas 2016, 45, 986-991, 10.1097/mpa.0000000000000590.
- Christie Y. Jeon; Sungjin Kim; Yu-Chen Lin; Harvey A. Risch; Mark O. Goodarzi; Teryl K. Nuckols; Stephen J. Freedland; Stephen J. Pandol; Joseph R. Pisegna; Prediction of Pancreatic Cancer in Diabetes Patients with Worsening Glycemic Control. Cancer Epidemiology, Biomarkers & Prevention 2022, 31, 242-253, 10.1158/1055-9965.epi-21-0712.
- Michael Bodmer; Claudia Becker; Christian Meier; Susan Jick; Christoph R Meier; Use of Antidiabetic Agents and the Risk of Pancreatic Cancer: A Case–Control Analysis. American Journal of Gastroenterology 2012, 107, 620-626, 10.1038/ajg.2011.483.
- I.N. Colmers; S.L. Bowker; L.A. Tjosvold; J.A. Johnson; Insulin use and cancer risk in patients with type 2 diabetes: A systematic review and meta-analysis of observational studies. Diabetes & Metabolism 2012, 38, 485-506, 10.1016/j.diabet.2012.08.011.
- Ali Vaziri-Gohar; Mahsa Zarei; Jonathan R. Brody; Jordan M. Winter; Metabolic Dependencies in Pancreatic Cancer. Frontiers in Oncology 2018, 8, 617, 10.3389/fonc.2018.00617.
- Chelsea Schiliro; Bonnie Firestein; Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056, 10.3390/cells10051056.
- Douglas E. Biancur; Alec C. Kimmelman; The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochimica et Biophysica Acta 2018, 1870, 67-75, 10.1016/j.bbcan.2018.04.011.
- Cristovão M. Sousa; Douglas E. Biancur; Xiaoxu Wang; Christopher J. Halbrook; Mara H. Sherman; Li Zhang; Daniel Kremer; Rosa F. Hwang; Agnes K. Witkiewicz; Haoqiang Ying; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature Cell Biology 2016, 536, 479-483, 10.1038/nature19084.
- Ramiz Ahmad; Timothy Eubank; Slawomir Lukomski; Brian Boone; Immune Cell Modulation of the Extracellular Matrix Contributes to the Pathogenesis of Pancreatic Cancer. Biomolecules 2021, 11, 901, 10.3390/biom11060901.
- Anneleen Daemen; David Peterson; Nisebita Sahu; Ron McCord; Xiangnan Du; Bonnie Liu; Katarzyna Kowanetz; Rebecca Hong; John Moffat; Min Gao; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proceedings of the National Academy of Sciences 2015, 112, E4410-E4417, 10.1073/pnas.1501605112.
- Jiyeon Kim; Ralph J. DeBerardinis; Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metabolism 2019, 30, 434-446, 10.1016/j.cmet.2019.08.013.
- Vincent Bernard; Alexander Semaan; Jonathan Huang; F. Anthony San Lucas; Feven C. Mulu; Bret M. Stephens; Paola A. Guerrero; Yanqing Huang; Jun Zhao; Nabiollah Kamyabi; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clinical Cancer Research 2019, 25, 2194-2205, 10.1158/1078-0432.ccr-18-1955.
- Junya Peng; Bao-Fa Sun; Chuan-Yuan Chen; Jia-Yi Zhou; Yu-Sheng Chen; Hao Chen; Lulu Liu; Dan Huang; Jialin Jiang; Guan-Shen Cui; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Research 2019, 29, 725-738, 10.1038/s41422-019-0195-y.
- Simone Hausmann; Bo Kong; Christoph Michalski; Mert Erkan; Helmut Friess; The Role of Inflammation in Pancreatic Cancer. null 2014, 816, 129-151, 10.1007/978-3-0348-0837-8_6.
- Eva M. Pålsson-McDermott; Luke A. J. O’Neill; Targeting immunometabolism as an anti-inflammatory strategy. Cell Research 2020, 30, 300-314, 10.1038/s41422-020-0291-z.
- Lidong Cao; Jiacheng Wu; Xianzhi Qu; Jiyao Sheng; Mengying Cui; Shui Liu; Xu Huang; Yien Xiang; Bingjin Li; Xuewen Zhang; et al. Glycometabolic rearrangements--aerobic glycolysis in pancreatic cancer: causes, characteristics and clinical applications. Journal of Experimental & Clinical Cancer Research 2020, 39, 1-22, 10.1186/s13046-020-01765-x.
- Sarah Davidson; Mirjana Efremova; Angela Riedel; Bidesh Mahata; Jhuma Pramanik; Jani Huuhtanen; Gozde Kar; Roser Vento-Tormo; Tzachi Hagai; Xi Chen; et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Reports 2020, 31, 107628, 10.1016/j.celrep.2020.107628.
- Nadine Assmann; David K. Finlay; Metabolic regulation of immune responses: therapeutic opportunities. Journal of Clinical Investigation 2016, 126, 2031-2039, 10.1172/jci83005.
- Hweixian Penny; Je Sieow; Sin Gun; Mai Lau; Bernett Lee; Jasmine Tan; Cindy Phua; Florida Toh; Yvonne Nga; Wei Yeap; et al. Targeting Glycolysis in Macrophages Confers Protection Against Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences 2021, 22, 6350, 10.3390/ijms22126350.
- Nikita Sharma; Vineet K. Gupta; Vanessa T. Garrido; Roey Hadad; Brittany C. Durden; Kousik Kesh; Bhuwan Giri; Anthony Ferrantella; Vikas Dudeja; Ashok Saluja; et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. Journal of Clinical Investigation 2019, 130, 451-465, 10.1172/jci127515.
- Haoqiang Ying; Alec C. Kimmelman; Costas A. Lyssiotis; Sujun Hua; Gerald C. Chu; Eliot Fletcher-Sananikone; Jason W. Locasale; Jaekyoung Son; Hailei Zhang; Jonathan L. Coloff; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656-670, 10.1016/j.cell.2012.01.058.
- Jihye Yun; Carlo Rago; Ian Cheong; Ray Pagliarini; Philipp Angenendt; Harith Rajagopalan; Kerstin Schmidt; James K. V. Willson; Sandy Markowitz; Shibin Zhou; et al. Glucose Deprivation Contributes to the Development of KRAS Pathway Mutations in Tumor Cells. Science 2009, 325, 1555-1559, 10.1126/science.1174229.
- Allison N Lau; Zhaoqi Li; Laura V Danai; Anna M Westermark; Alicia M Darnell; Raphael Ferreira; Vasilena Gocheva; Sharanya Sivanand; Evan C Lien; Kiera M Sapp; et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. eLife 2020, 9, 234-239, 10.7554/elife.56782.
- Hiroshi Kurahara; Kosei Maemura; Yuko Mataki; Masahiko Sakoda; Satoshi Iino; Yota Kawasaki; Takaaki Arigami; Shinichiro Mori; Yuko Kijima; Shinichi Ueno; et al. Significance of Glucose Transporter Type 1 (GLUT-1) Expression in the Therapeutic Strategy for Pancreatic Ductal Adenocarcinoma. Annals of Surgical Oncology 2018, 25, 1432-1439, 10.1245/s10434-018-6357-1.
- Kinga B. Stopa; Agnieszka A. Kusiak; Mateusz D. Szopa; Pawel E. Ferdek; Monika A. Jakubowska; Pancreatic Cancer and Its Microenvironment—Recent Advances and Current Controversies. International Journal of Molecular Sciences 2020, 21, 3218, 10.3390/ijms21093218.
- Thomas R. Cox; The matrix in cancer. Nature Cancer 2021, 21, 217-238, 10.1038/s41568-020-00329-7.
- Minoti V. Apte; Jeremy S. Wilson; Aurelia Lugea; Stephen J. Pandol; A Starring Role for Stellate Cells in the Pancreatic Cancer Microenvironment. Gastroenterology 2013, 144, 1210-1219, 10.1053/j.gastro.2012.11.037.
- M Sinn; Carsten Denkert; J K Striefler; U Pelzer; J M Stieler; M Bahra; P Lohneis; B Dörken; H Oettle; H Riess; et al. α-Smooth muscle actin expression and desmoplastic stromal reaction in pancreatic cancer: results from the CONKO-001 study. British Journal of Cancer 2014, 111, 1917-1923, 10.1038/bjc.2014.495.
- Vegard Tjomsland; Anna Spångeus; Johanna Välilä; Per Sandström; Kurt Borch; Henrik Druid; Sture Falkmer; Ursula Falkmer; Davorka Messmer; Marie Larsson; et al. Interleukin 1α Sustains the Expression of Inflammatory Factors in Human Pancreatic Cancer Microenvironment by Targeting Cancer-Associated Fibroblasts. Neoplasia 2011, 13, 664-IN3, 10.1593/neo.11332.
- Zhuonan Zhuang; Huai-Qiang Ju; Mitzi Aguilar; Takashi Gocho; Hao Li; Tomonori Iida; Harold Lee; Xiaoqiang Fan; Haijun Zhou; Jianhua Ling; et al. IL1 Receptor Antagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-κB Activation. Clinical Cancer Research 2016, 22, 1432-1444, 10.1158/1078-0432.ccr-14-3382.
- Giulia Biffi; Tobiloba E. Oni; Benjamin Spielman; Yuan Hao; Ela Elyada; Youngkyu Park; Jonathan Preall; David A. Tuveson; IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discovery 2019, 9, 282-301, 10.1158/2159-8290.cd-18-0710.
- Veli-Matti Leppänen; Pipsa Saharinen; Kari Alitalo; Structural basis of Tie2 activation and Tie2/Tie1 heterodimerization. Proceedings of the National Academy of Sciences 2017, 114, 4376-4381, 10.1073/pnas.1616166114.
- Emilia A. Korhonen; Anita Lampinen; Hemant Giri; Andrey Anisimov; Minah Kim; Breanna Allen; Shentong Fang; Gabriela D’Amico; Tuomas J. Sipilä; Marja Lohela; et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. Journal of Clinical Investigation 2016, 126, 3495-3510, 10.1172/jci84923.
- Masahiro Inoue; Jeffrey H Hager; Napoleone Ferrara; Hans-Peter Gerber; Douglas Hanahan; VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic β cell carcinogenesis. Cancer Cell 2002, 1, 193-202, 10.1016/s1535-6108(02)00031-4.
- Vito Longo; Oronzo Brunetti; Antonio Gnoni; Stefano Cascinu; Giampietro Gasparini; Vito Lorusso; Domenico Ribatti; Nicola Silvestris; Angiogenesis in pancreatic ductal adenocarcinoma: A controversial issue. Oncotarget 2016, 7, 58649-58658, 10.18632/oncotarget.10765.
- Panagiotis Sarantis; Evangelos Koustas; Adriana Papadimitropoulou; Athanasios G Papavassiliou; Michalis V Karamouzis; Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World Journal of Gastrointestinal Oncology 2020, 12, 173-181, 10.4251/wjgo.v12.i2.173.
- Bolun Jiang; Li Zhou; Jun Lu; Yizhi Wang; Chengxi Liu; Lei You; Junchao Guo; Stroma-Targeting Therapy in Pancreatic Cancer: One Coin With Two Sides?. Frontiers in Oncology 2020, 10, 576399, 10.3389/fonc.2020.576399.
- Jessica K. Nelson; May Zaw Thin; Theodore Evan; Steven Howell; Mary Wu; Bruna Almeida; Nathalie Legrave; Duco S. Koenis; Gabriela Koifman; Yoichiro Sugimoto; et al. USP25 promotes pathological HIF-1-driven metabolic reprogramming and is a potential therapeutic target in pancreatic cancer. Nature Communications 2022, 13, 1-18, 10.1038/s41467-022-29684-9.
- Claudio Scafoglio; Bruce A. Hirayama; Vladimir Kepe; Jie Liu; Chiara Ghezzi; Nagichettiar Satyamurthy; Neda A. Moatamed; Jiaoti Huang; Hermann Koepsell; Jorge R. Barrio; et al. Functional expression of sodium-glucose transporters in cancer. Proceedings of the National Academy of Sciences 2015, 112, E4111-E4119, 10.1073/pnas.1511698112.
- Felipe Paredes; Holly C. Williams; Alejandra San Martin; Metabolic adaptation in hypoxia and cancer. Cancer Letters 2021, 502, 133-142, 10.1016/j.canlet.2020.12.020.
- Siyuan Chen; Bo Ning; Jinwen Song; Zihan Yang; Li Zhou; Zhiji Chen; Linhong Mao; Hongtao Liu; Qingliang Wang; Song He; et al. Enhanced pentose phosphate pathway activity promotes pancreatic ductal adenocarcinoma progression via activating YAP/MMP1 axis under chronic acidosis. International Journal of Biological Sciences 2022, 18, 2304-2316, 10.7150/ijbs.69526.
- Su Chii Kong; Asbjørn Nøhr-Nielsen; Katrine Zeeberg; Stephan Joel Reshkin; Else Kay Hoffmann; Ivana Novak; Stine Falsig Pedersen; Monocarboxylate Transporters MCT1 and MCT4 Regulate Migration and Invasion of Pancreatic Ductal Adenocarcinoma Cells. Pancreas 2016, 45, 1036-1047, 10.1097/mpa.0000000000000571.
- GuemHee Baek; Yan F. Tse; Zeping Hu; Derek Cox; Noah Buboltz; Peter McCue; Charles J. Yeo; Michael A. White; Ralph J. DeBerardinis; Erik S. Knudsen; et al. MCT4 Defines a Glycolytic Subtype of Pancreatic Cancer with Poor Prognosis and Unique Metabolic Dependencies. Cell Reports 2014, 9, 2233-2249, 10.1016/j.celrep.2014.11.025.
- William J. Quinn; Jing Jiao; Tara TeSlaa; Jason Stadanlick; Zhonglin Wang; Liqing Wang; Tatiana Akimova; Alessia Angelin; Patrick M. Schäfer; Michelle D. Cully; et al. Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Reports 2020, 33, 108500-108500, 10.1016/j.celrep.2020.108500.
- Maria V. Liberti; Jason W. Locasale; The Warburg Effect: How Does it Benefit Cancer Cells?. Trends in Biochemical Sciences 2016, 41, 211-218, 10.1016/j.tibs.2015.12.001.
- Surendra K. Shukla; Vinee Purohit; Kamiya Mehla; Venugopal Gunda; Nina V. Chaika; Enza Vernucci; Ryan J. King; Jaime Abrego; Gennifer D. Goode; Aneesha Dasgupta; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 392, 10.1016/j.ccell.2017.08.008.
- Sabine Riethdorf; Natalie Reimers; Volker Assmann; Jan-Wilhelm Kornfeld; Luigi Terracciano; Guido Sauter; Klaus Pantel; High incidence of EMMPRIN expression in human tumors. International Journal of Cancer 2006, 119, 1800-1810, 10.1002/ijc.22062.
- P. Kirk; M.C. Wilson; C. Heddle; M.H. Brown; A.N. Barclay; A.P. Halestrap; CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. The EMBO Journal 2000, 19, 3896-3904, 10.1093/emboj/19.15.3896.
- W Schneiderhan; M Scheler; K-H Holzmann; M Marx; J E Gschwend; M Bucholz; Thomas Gress; T Seufferlein; G Adler; F Oswald; et al. CD147 silencing inhibits lactate transport and reduces malignant potential of pancreatic cancer cells in in vivo and in vitro models. Gut 2009, 58, 1391-1398, 10.1136/gut.2009.181412.
- Krushna C. Patra; Nissim Hay; The pentose phosphate pathway and cancer. Trends in Biochemical Sciences 2014, 39, 347-354, 10.1016/j.tibs.2014.06.005.
- Matthew E. Bechard; Anna Word; Amanda V. Tran; Xiaojing Liu; Jason W. Locasale; Oliver G. McDonald; Pentose conversions support the tumorigenesis of pancreatic cancer distant metastases. Oncogene 2018, 37, 5248-5256, 10.1038/s41388-018-0346-5.
- Nicholas J Kruger; Antje von Schaewen; The oxidative pentose phosphate pathway: structure and organisation. Current Opinion in Plant Biology 2003, 6, 236-246, 10.1016/s1369-5266(03)00039-6.
- Anna Stincone; Alessandro Prigione; Thorsten Cramer; Mirjam M. C. Wamelink; Kate Campbell; Eric Cheung; Viridiana Olin‐Sandoval; Nana-Maria Greuning; Antje Krueger; Mohammad Tauqeer Alam; et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biological Reviews 2014, 90, 927-963, 10.1111/brv.12140.
- Naiara Santana Codina; Anjali A. Roeth; Yi Zhang; Annan Yang; Oksana Mashadova; John M. Asara; Xiaoxu Wang; Roderick T. Bronson; Costas A. Lyssiotis; Haoqiang Ying; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nature Communications 2018, 9, 1-13, 10.1038/s41467-018-07472-8.
- Ying Yang; Mari B. Ishak Gabra; Eric A. Hanse; Xazmin H. Lowman; Thai Q. Tran; Haiqing Li; Neta Milman; Juan Liu; Michael Reid; Jason W. Locasale; et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nature Communications 2019, 10, 1-15, 10.1038/s41467-019-08759-0.
- De-Hai Wu; Hao Liang; Shou-Nan Lu; Hao Wang; Zhi-Lei Su; Lei Zhang; Jian-Qun Ma; Mian Guo; Sheng Tai; Shan Yu; et al. miR-124 Suppresses Pancreatic Ductal Adenocarcinoma Growth by Regulating Monocarboxylate Transporter 1-Mediated Cancer Lactate Metabolism. Cellular Physiology and Biochemistry 2018, 50, 924-935, 10.1159/000494477.
- Makoto Abue; Misa Yokoyama; Rie Shibuya; Keiichi Tamai; Kazunori Yamaguchi; Ikuro Sato; Nobuyuki Tanaka; Shin Hamada; Tooru Shimosegawa; Kazuo Sugamura; et al. Circulating miR-483-3p and miR-21 is highly expressed in plasma of pancreatic cancer. International Journal of Oncology 2014, 46, 539-547, 10.3892/ijo.2014.2743.
- Afra Z. Daoud; Eoghan Mulholland; Grace Cole; Helen O. McCarthy; MicroRNAs in Pancreatic Cancer: biomarkers, prognostic, and therapeutic modulators. BMC Cancer 2019, 19, 1-13, 10.1186/s12885-019-6284-y.
- Ruiyuan Xu; Jinshou Yang; Bo Ren; Huanyu Wang; Gang Yang; Yuan Chen; Lei You; Yupei Zhao; Reprogramming of Amino Acid Metabolism in Pancreatic Cancer: Recent Advances and Therapeutic Strategies. Frontiers in Oncology 2020, 10, 572722, 10.3389/fonc.2020.572722.
- Vinicius Cruzat; Marcelo Macedo Rogero; Kevin Noel Keane; Rui Curi; Philip Newsholme; Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564, 10.3390/nu10111564.
- Brian J. Altman; Zachary E. Stine; Chi V. Dang; From Krebs to clinic: glutamine metabolism to cancer therapy. Nature Cancer 2016, 16, 619-634, 10.1038/nrc.2016.71.
- Ahmad A. Cluntun; Michael J. Lukey; Richard A. Cerione; Jason W. Locasale; Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends in Cancer 2017, 3, 169-180, 10.1016/j.trecan.2017.01.005.
- Jurre J. Kamphorst; Michel Nofal; Cosimo Commisso; Sean R. Hackett; Wenyun Lu; Elda Grabocka; Matthew G. Vander Heiden; George Miller; Jeffrey A. Drebin; Dafna Bar-Sagi; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Research 2015, 75, 544-553, 10.1158/0008-5472.can-14-2211.
- Alex J. Bott; Jianliang Shen; Claudia Tonelli; Le Zhan; Nithya Sivaram; Ya-Ping Jiang; Xufen Yu; Vrushank Bhatt; Eric Chiles; Hua Zhong; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Reports 2019, 29, 1287-1298.e6, 10.1016/j.celrep.2019.09.056.
- Gabriela Reyes-Castellanos; Nadine Abdel Hadi; Alice Carrier; Autophagy Contributes to Metabolic Reprogramming and Therapeutic Resistance in Pancreatic Tumors. Cells 2022, 11, 426, 10.3390/cells11030426.
- Chia-Jung Li; Wan-Ting Liao; Meng-Yu Wu; Pei-Yi Chu; New Insights into the Role of Autophagy in Tumor Immune Microenvironment. International Journal of Molecular Sciences 2017, 18, 1566, 10.3390/ijms18071566.
- Cosimo Commisso; Shawn M. Davidson; Rengin G. Soydaner-Azeloglu; Seth J. Parker; Jurre J. Kamphorst; Sean Hackett; Elda Grabocka; Michel Nofal; Jeffrey A. Drebin; Craig B. Thompson; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633-637, 10.1038/nature12138.
- David R. Wise; Ralph J. DeBerardinis; Anthony Mancuso; Nabil Sayed; Xiao-Yong Zhang; Harla K. Pfeiffer; Ilana Nissim; Evgueni Daikhin; Marc Yudkoff; Steven B. McMahon; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences 2008, 105, 18782-18787, 10.1073/pnas.0810199105.
- Dafna Bar-Sagi; James R. Feramisco; Induction of Membrane Ruffling and Fluid-Phase Pinocytosis in Quiescent Fibroblasts by ras Proteins. Science 1986, 233, 1061-1068, 10.1126/science.3090687.
- Natalie Porat-Shliom; Yoel Kloog; Julie G. Donaldson; A Unique Platform for H-Ras Signaling Involving Clathrin-independent Endocytosis. Molecular Biology of the Cell 2008, 19, 765-775, 10.1091/mbc.e07-08-0841.
- Ju-Won Seo; Jungwon Choi; So-Yeon Lee; Suhyun Sung; Hyun Ju Yoo; Min-Ji Kang; Heesun Cheong; Jaekyoung Son; Autophagy is required for PDAC glutamine metabolism. Scientific Reports 2016, 6, 37594, 10.1038/srep37594.
- Bryce W. Carey; Lydia W. S. Finley; Justin R. Cross; C. David Allis; Craig B. Thompson; Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2014, 518, 413-416, 10.1038/nature13981.
- Thai Q. Tran; Mari B. Ishak Gabra; Xazmin H. Lowman; Ying Yang; Michael Reid; Min Pan; Timothy R. O’Connor; Mei Kong; Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLOS Biology 2017, 15, e2002810-e2002810, 10.1371/journal.pbio.2002810.
- Seth J. Parker; Caroline R. Amendola; Kate E. R. Hollinshead; Qijia Yu; Keisuke Yamamoto; Joel Encarnación-Rosado; Rebecca E. Rose; Madeleine M. LaRue; Albert S. W. Sohn; Doug E. Biancur; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discovery 2020, 10, 1018-1037, 10.1158/2159-8290.cd-19-0959.
- Nadine Sperb; Miltiadis Tsesmelis; Thomas Wirth; Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences 2020, 21, 5486, 10.3390/ijms21155486.
- Jakob Michael Riedl; Florian Posch; Gerald Prager; Wolfgang Eisterer; Leopold Oehler; Thamer Sliwa; Klaus Wilthoner; Andreas Petzer; Petra Pichler; Eva Hubmann; et al. The AST/ALT (De Ritis) ratio predicts clinical outcome in patients with pancreatic cancer treated with first-line nab-paclitaxel and gemcitabine: post hoc analysis of an Austrian multicenter, noninterventional study. Therapeutic Advances in Medical Oncology 2020, 12, 1243, 10.1177/1758835919900872.
- Orianne Olivares; Jared R. Mayers; Victoire Gouirand; Margaret E. Torrence; Tristan Gicquel; Laurence Borge; Sophie Lac; Julie Roques; Marie-Noëlle Lavaut; Patrice Berthezène; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nature Communications 2017, 8, 16031-16031, 10.1038/ncomms16031.
- Mylène Tajan; Andreas K. Hock; Julianna Blagih; Neil A. Robertson; Christiaan F. Labuschagne; Flore Kruiswijk; Timothy J. Humpton; Peter D. Adams; Karen H. Vousden; A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metabolism 2018, 28, 721-736.e6, 10.1016/j.cmet.2018.07.005.
- Ru Chen; Lisa A Lai; Yumi Sullivan; Melissa Wong; Lei Wang; Jonah Riddell; Linda Jung; Venu G. Pillarisetty; Teresa A. Brentnall; Sheng Pan; et al. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Scientific Reports 2017, 7, 1-14, 10.1038/s41598-017-08436-6.
- Claudia C.S. Chini; Anatilde M. Gonzalez Guerrico; Veronica Nin; Juliana Camacho-Pereira; Carlos Escande; Maria Thereza Barbosa; Eduardo N. Chini; Targeting of NAD Metabolism in Pancreatic Cancer Cells: Potential Novel Therapy for Pancreatic Tumors. Clinical Cancer Research 2014, 20, 120-130, 10.1158/1078-0432.ccr-13-0150.
- Oliver Maddocks; Dimitris Athineos; Eric C. Cheung; Pearl Lee; Tong Zhang; Niels J. F. Van Den Broek; Gillian M. Mackay; Christiaan F. Labuschagne; David Michael Gay; Flore Kruiswijk; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372-376, 10.1038/nature22056.
- Michael A. Badgley; Daniel M. Kremer; H. Carlo Maurer; Kathleen E. DelGiorno; Ho-Joon Lee; Vinee Purohit; Irina R. Sagalovskiy; Alice Ma; Jonathan Kapilian; Christina E. M. Firl; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85-89, 10.1126/science.aaw9872.
- Marco E. Bianchi; DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of Leukocyte Biology 2006, 81, 1-5, 10.1189/jlb.0306164.
- J. M. Genkinger; C. M. Kitahara; L. Bernstein; A. Berrington de Gonzalez; M. Brotzman; J. W. Elena; G. G. Giles; P. Hartge; P. N. Singh; R. Z. Stolzenberg-Solomon; et al. Central adiposity, obesity during early adulthood, and pancreatic cancer mortality in a pooled analysis of cohort studies. Annals of Oncology 2015, 26, 2257-2266, 10.1093/annonc/mdv355.
- Tobiloba E. Oni; Giulia Biffi; Lindsey A. Baker; Yuan Hao; Claudia Tonelli; Tim D.D. Somerville; Astrid Deschênes; Pascal Belleau; Chang-Il Hwang; Francisco J. Sánchez-Rivera; et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. Journal of Experimental Medicine 2020, 217, 321, 10.1084/jem.20192389.
- Linara Gabitova-Cornell; Aizhan Surumbayeva; Suraj Peri; Janusz Franco-Barraza; Diana Restifo; Nicole Weitz; Charline Ogier; Aaron R. Goldman; Tiffiney R. Hartman; Ralph Francescone; et al. Cholesterol Pathway Inhibition Induces TGF-β Signaling to Promote Basal Differentiation in Pancreatic Cancer. Cancer Cell 2020, 38, 567-583.e11, 10.1016/j.ccell.2020.08.015.
- Sandrine Silvente-Poirot; Marc Poirot; Cholesterol and Cancer, in the Balance. Science 2014, 343, 1445-1446, 10.1126/science.1252787.
- P Clerc; N Bensaadi; P Pradel; A Estival; F Clemente; N Vaysse; Lipid-dependent proliferation of pancreatic cancer cell lines.. Cancer Research 1991, 51, 435, .
- Julian Swierczynski; Areta Hebanowska; Tomasz Sledzinski; Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World Journal of Gastroenterology 2014, 20, 725, 10.3748/wjg.v20.i9.2279.
- Kim Walter; Seung-Mo Hong; Sinead Nyhan; Marcia Canto; Neal Fedarko; Alison Klein; Margaret Griffith; Noriyuki Omura; Susan Medghalchi; Frank Kuhajda; et al. Serum Fatty Acid Synthase as a Marker of Pancreatic Neoplasia. Cancer Epidemiology, Biomarkers & Prevention 2009, 18, 2380-2385, 10.1158/1055-9965.epi-09-0144.
- Yan Sun; Weiwei He; Man Luo; Yuhong Zhou; Guilin Chang; Weiying Ren; Kefen Wu; Chang Guilin; Jiping Shen; Xiaoping Zhao; et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumor Biology 2015, 36, 4133-4141, 10.1007/s13277-015-3047-5.
- Saber Tadros; Surendra K. Shukla; Ryan J. King; Venugopal Gunda; Enza Vernucci; Jaime Abrego; Nina V. Chaika; Fang Yu; Audrey J. Lazenby; Lyudmyla Berim; et al. De Novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Research 2017, 77, 5503-5517, 10.1158/0008-5472.can-16-3062.
- Youyun Yang; Hailan Li; Zhaomin Li; Zijin Zhao; Michelle Yip-Schneider; Qipeng Fan; C. Max Schmidt; E. Gabriela Chiorean; Jingwu Xie; Liang Cheng; et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. International journal of biochemistry and molecular biology 2011, 2, 89-98, .
- Cody N. Rozeveld; Katherine M. Johnson; Lizhi Zhang; Gina L. Razidlo; KRAS Controls Pancreatic Cancer Cell Lipid Metabolism and Invasive Potential through the Lipase HSL. Cancer Research 2020, 80, 4932-4945, 10.1158/0008-5472.can-20-1255.
- Knut Tomas Dalen; Stine M. Ulven; Borghild M. Arntsen; Karianne Solaas; Hilde I. Nebb; PPARα activators and fasting induce the expression of adipose differentiation-related protein in liver. Journal of Lipid Research 2006, 47, 931-943, 10.1194/jlr.m500459-jlr200.
- Benny Hung-Junn Chang; Lan Li; Antoni Paul; Susumu Taniguchi; Vijayalakshmi Nannegari; William C. Heird; Lawrence Chan; Protection against Fatty Liver but Normal Adipogenesis in Mice Lacking Adipose Differentiation-Related Protein. Molecular and Cellular Biology 2006, 26, 1063-1076, 10.1128/mcb.26.3.1063-1076.2006.
- Yuki Hashimoto; Mitsuaki Ishida; Hironori Ryota; Tomohisa Yamamoto; Hisashi Kosaka; Satoshi Hirooka; So Yamaki; Masaya Kotsuka; Yoichi Matsui; Hiroaki Yanagimoto; et al. Adipophilin expression is an indicator of poor prognosis in patients with pancreatic ductal adenocarcinoma: An immunohistochemical analysis. Pancreatology 2019, 19, 443-448, 10.1016/j.pan.2019.03.001.
- Elaine Chen; Tsung Huang Tsai; Lan Li; Pradip Saha; Lawrence Chan; Benny Hung-Junn Chang; PLIN2 is a Key Regulator of the Unfolded Protein Response and Endoplasmic Reticulum Stress Resolution in Pancreatic β Cells. Scientific Reports 2017, 7, 40855, 10.1038/srep40855.
- Susanne M. Schmidt; Kerstin Schag; Martin R. Müller; Toni Weinschenk; Silke Appel; Oliver Schoor; Markus M. Weck; Frank Grünebach; Lothar Kanz; Stefan Stevanovic; et al. Induction of Adipophilin-Specific Cytotoxic T Lymphocytes Using a Novel HLA-A2-Binding Peptide That Mediates Tumor Cell Lysis. Cancer Research 2004, 64, 1164-1170, 10.1158/0008-5472.can-03-2538.
- Adrian M. Seifert; Charlotte Reiche; Max Heiduk; Anna Tannert; Ann-Christin Meinecke; Stephanie Baier; Janusz von Renesse; Christoph Kahlert; Marius Distler; Thilo Welsch; et al. Detection of pancreatic ductal adenocarcinoma with galectin-9 serum levels. Oncogene 2020, 39, 3102-3113, 10.1038/s41388-020-1186-7.
- Yijia He; Yuexin Dong; Xinwen Zhang; Zhuang Ding; Yuxian Song; Xiaofeng Huang; Sheng Chen; Zhiyong Wang; Yanhong Ni; Liang Ding; et al. Lipid Droplet-Related PLIN2 in CD68+ Tumor-Associated Macrophage of Oral Squamous Cell Carcinoma: Implications for Cancer Prognosis and Immunotherapy. Frontiers in Oncology 2022, 12, 287, 10.3389/fonc.2022.824235.
- Bincy Philip; Christina L. Roland; Jaroslaw Daniluk; Yan Liu; Deyali Chatterjee; Sobeyda B. Gomez; Baoan Ji; Haojie Huang; Huamin Wang; Jason B. Fleming; et al. A High-Fat Diet Activates Oncogenic Kras and COX2 to Induce Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2013, 145, 1449-1458, 10.1053/j.gastro.2013.08.018.
- Katherine Minjee Chung; Jaffarguriqbal Singh; Lauren Lawres; Kimberly Judith Dorans; Cathy Garcia; Daniel Burkhardt; Rebecca Robbins; Arjun Bhutkar; Rebecca Cardone; Xiaojian Zhao; et al. Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma. Cell 2020, 181, 832-847.e18, 10.1016/j.cell.2020.03.062.
- Andrew S. Greenberg; Rosalind A. Coleman; Fredric B. Kraemer; James L. McManaman; Martin S. Obin; Vishwajeet Puri; Qing-Wu Yan; Hideaki Miyoshi; Douglas G. Mashek; The role of lipid droplets in metabolic disease in rodents and humans. Journal of Clinical Investigation 2011, 121, 2102-2110, 10.1172/jci46069.
- James A. Olzmann; Pedro Carvalho; Dynamics and functions of lipid droplets. Nature Reviews Molecular Cell Biology 2018, 20, 137-155, 10.1038/s41580-018-0085-z.
- Fabienne Guillaumond; Ghislain Bidaut; Mehdi Ouaissi; Stéphane Servais; Victoire Gouirand; Orianne Olivares; Sophie Lac; Laurence Borge; Julie Roques; Odile Gayet; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proceedings of the National Academy of Sciences 2015, 112, 2473-2478, 10.1073/pnas.1421601112.
- Jurre J. Kamphorst; Justin R. Cross; Jing Fan; Elisa de Stanchina; Robin Mathew; Eileen P. White; Craig B. Thompson; Joshua D. Rabinowitz; Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proceedings of the National Academy of Sciences 2013, 110, 8882-8887, 10.1073/pnas.1307237110.
- Mahesh S. Padanad; Georgia Konstantinidou; Niranjan Venkateswaran; Margherita Melegari; Smita Rindhe; Matthew Mitsche; Chendong Yang; Kimberly Batten; Kenneth E. Huffman; Jingwen Liu; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Reports 2016, 16, 1614-1628, 10.1016/j.celrep.2016.07.009.
- Frits Kamp; James Hamilton; How fatty acids of different chain length enter and leave cells by free diffusion. Prostaglandins, Leukotrienes and Essential Fatty Acids 2006, 75, 149-159, 10.1016/j.plefa.2006.05.003.
- Matteo Rossi Sebastiano; Georgia Konstantinidou; Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. International Journal of Molecular Sciences 2019, 20, 3624, 10.3390/ijms20153624.
- Maria Saliakoura; Matteo Rossi Sebastiano; Ioanna Nikdima; Chiara Pozzato; Georgia Konstantinidou; Restriction of extracellular lipids renders pancreatic cancer dependent on autophagy. Journal of Experimental & Clinical Cancer Research 2022, 41, 1-13, 10.1186/s13046-021-02231-y.
- Jessica Brandi; Ilaria Dando; Elisa Dalla Pozza; Giulia Biondani; Rosalind Jenkins; Victoria Elliott; Kevin Park; Giuseppina Fanelli; Lello Zolla; Eithne Costello; et al. Proteomic analysis of pancreatic cancer stem cells: Functional role of fatty acid synthesis and mevalonate pathways. Journal of Proteomics 2017, 150, 310-322, 10.1016/j.jprot.2016.10.002.
- Claudia Di Carlo; Bebiana C. Sousa; Marcello Manfredi; Jessica Brandi; Elisa Dalla Pozza; Emilio Marengo; Marta Palmieri; Ilaria Dando; Michael J. O. Wakelam; Andrea F. Lopez-Clavijo; et al. Integrated lipidomics and proteomics reveal cardiolipin alterations, upregulation of HADHA and long chain fatty acids in pancreatic cancer stem cells. Scientific Reports 2021, 11, 1-13, 10.1038/s41598-021-92752-5.
- Mohammed El-Hafidi; Francisco Correa; Cecilia Zazueta; Mitochondrial dysfunction in metabolic and cardiovascular diseases associated with cardiolipin remodeling. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2020, 1866, 165744, 10.1016/j.bbadis.2020.165744.
- Kathy Pfeiffer; Vishal Gohil; Rosemary A. Stuart; Carola Hunte; Ulrich Brandt; Miriam L. Greenberg; Hermann Schägger; Cardiolipin Stabilizes Respiratory Chain Supercomplexes. Journal of Biological Chemistry 2003, 278, 52873-52880, 10.1074/jbc.m308366200.
- Denise Wolrab; Robert Jirásko; Eva Cífková; Marcus Höring; Ding Mei; Michaela Chocholoušková; Ondřej Peterka; Jakub Idkowiak; Tereza Hrnčiarová; Ladislav Kuchař; et al. Lipidomic profiling of human serum enables detection of pancreatic cancer. Nature Communications 2022, 13, 1-16, 10.1038/s41467-021-27765-9.
- Julia Mayerle; Holger Kalthoff; Regina Reszka; Beate Kamlage; Erik Peter; Bodo Schniewind; Sandra González Maldonado; Christian Pilarsky; Claus-Dieter Heidecke; Philipp Schatz; et al. Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut 2017, 67, 128-137, 10.1136/gutjnl-2016-312432.
- Johannes F Fahrmann; Leonidas E Bantis; Michela Capello; Ghislaine Scelo; Jennifer B Dennison; Nikul Patel; Eunice Murage; Jody Vykoukal; Deepali L Kundnani; Lenka Foretova; et al. A Plasma-Derived Protein-Metabolite Multiplexed Panel for Early-Stage Pancreatic Cancer. JNCI: Journal of the National Cancer Institute 2018, 111, 372-379, 10.1093/jnci/djy126.
- Guangxi Wang; Hantao Yao; Yan Gong; Zipeng Lu; Ruifang Pang; Yang Li; Yuyao Yuan; Huajie Song; Jia Liu; Yan Jin; et al. Metabolic detection and systems analyses of pancreatic ductal adenocarcinoma through machine learning, lipidomics, and multi-omics. Science Advances 2021, 7, 456, 10.1126/sciadv.abh2724.
- Rocio I. R. Macias; Luis Muñoz-Bellvís; Anabel Sánchez-Martín; Enara Arretxe; Ibon Martínez-Arranz; Ainhoa Lapitz; M. Laura Gutiérrez; Adelaida La La Casta; Cristina Alonso; Luis M. González; et al. A Novel Serum Metabolomic Profile for the Differential Diagnosis of Distal Cholangiocarcinoma and Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 1433, 10.3390/cancers12061433.
- Sebastian Doll; Bettina Proneth; Yulia Tyurina; Elena Panzilius; Sho Kobayashi; Irina Ingold; Martin Irmler; Martin Irmler Johannes Beckers; Michaela Aichler; Michaela Aichler Axel Walch; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature Chemical Biology 2016, 13, 91-98, 10.1038/nchembio.2239.
- Nanjun Hu; Lulu Bai; Enyong Dai; Leng Han; Rui Kang; Hongjun Li; Daolin Tang; Pirin is a nuclear redox-sensitive modulator of autophagy-dependent ferroptosis. Biochemical and Biophysical Research Communications 2020, 536, 100-106, 10.1016/j.bbrc.2020.12.066.
- Takuma Okawa; Motoyoshi Nagai; Koji Hase; Dietary Intervention Impacts Immune Cell Functions and Dynamics by Inducing Metabolic Rewiring. Frontiers in Immunology 2021, 11, 342, 10.3389/fimmu.2020.623989.
- Róisín M. Loftus; David K. Finlay; Immunometabolism: Cellular Metabolism Turns Immune Regulator. Journal of Biological Chemistry 2016, 291, 1-10, 10.1074/jbc.r115.693903.
- Chih-Hao Chang; Jing Qiu; David O’Sullivan; Michael D. Buck; Takuro Noguchi; Jonathan D. Curtis; Qiongyu Chen; Mariel Gindin; Matthew M. Gubin; Gerritje J.W. van der Windt; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229-1241, 10.1016/j.cell.2015.08.016.
- Akiyoshi Hirayama; Kenjiro Kami; Masahiro Sugimoto; Maki Sugawara; Naoko Toki; Hiroko Onozuka; Taira Kinoshita; Norio Saito; Atsushi Ochiai; Masaru Tomita; et al. Quantitative Metabolome Profiling of Colon and Stomach Cancer Microenvironment by Capillary Electrophoresis Time-of-Flight Mass Spectrometry. Cancer Research 2009, 69, 4918-4925, 10.1158/0008-5472.can-08-4806.
- Atsuo Ochi; Andrew H. Nguyen; Andrea S. Bedrosian; Harry M. Mushlin; Saman Zarbakhsh; Rocky Barilla; Constantinos P. Zambirinis; Nina C. Fallon; Adeel Rehman; Yuliya Pylayeva-Gupta; et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. Journal of Experimental Medicine 2012, 209, 1671-1687, 10.1084/jem.20111706.
- Stephen Yiu Chuen Choi; Colin C Collins; Peter W Gout; Yuzhuo Wang; Cancer‐generated lactic acid: a regulatory, immunosuppressive metabolite?. The Journal of Pathology 2013, 230, 350-355, 10.1002/path.4218.
- Akira Fukunaga; Masaki Miyamoto; Yasushi Cho; Soichi Murakami; You Kawarada; Taro Oshikiri; Kentaro Kato; Takanori Kurokawa; Masato Suzuoki; Yoshihiro Nakakubo; et al. CD8+ Tumor-Infiltrating Lymphocytes Together with CD4+ Tumor-Infiltrating Lymphocytes and Dendritic Cells Improve the Prognosis of Patients with Pancreatic Adenocarcinoma. Pancreas 2004, 28, e26-e31, 10.1097/00006676-200401000-00023.
- Yoshinori Ino; R Yamazaki-Itoh; Kazuaki Shimada; M Iwasaki; Tomoo Kosuge; Yae Kanai; Nobuyoshi Hiraoka; Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. British Journal of Cancer 2013, 108, 914-923, 10.1038/bjc.2013.32.
- Ji Sung Kim; Yun Soo Park; Ju Young Kim; Yong Guk Kim; Yeon Jin Kim; Hong Kyung Lee; Hyung Sook Kim; Jin Tae Hong; Youngsoo Kim; Sang-Bae Han; et al. Inhibition of Human Pancreatic Tumor Growth by Cytokine-Induced Killer Cells in Nude Mouse Xenograft Model. Immune Network 2012, 12, 247-52, 10.4110/in.2012.12.6.247.
- Keisuke Yamamoto; Anthony Venida; Julian Yano; Douglas E. Biancur; Miwako Kakiuchi; Suprit Gupta; Albert S. W. Sohn; Subhadip Mukhopadhyay; Elaine Y. Lin; Seth Parker; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature Cell Biology 2020, 581, 100-105, 10.1038/s41586-020-2229-5.
- Li Liu; Guochao Zhao; Wenchuan Wu; Yefei Rong; Dayong Jin; Dansong Wang; Wenhui Lou; Xinyu Qin; Low intratumoral regulatory T cells and high peritumoral CD8+ T cells relate to long-term survival in patients with pancreatic ductal adenocarcinoma after pancreatectomy. Cancer Immunology, Immunotherapy 2015, 65, 73-82, 10.1007/s00262-015-1775-4.
- Christian Kurts; Th17 cells: a third subset of CD4+ T effector cells involved in organ-specific autoimmunity. Nephrology Dialysis Transplantation 2007, 23, 816-819, 10.1093/ndt/gfm800.
- S M Wörmann; K N Diakopoulos; M Lesina; H Algül; The immune network in pancreatic cancer development and progression. Oncogene 2013, 33, 2956-2967, 10.1038/onc.2013.257.
- Jennifer L. Gnerlich; Jonathan Mitchem; Joshua S. Weir; Narendra V. Sankpal; Hiroyuki Kashiwagi; Brian A. Belt; Matthew Porembka; John M. Herndon; Timothy J. Eberlein; Peter Goedegebuure; et al. Induction of Th17 Cells in the Tumor Microenvironment Improves Survival in a Murine Model of Pancreatic Cancer. The Journal of Immunology 2010, 185, 4063-4071, 10.4049/jimmunol.0902609.
- J. H. Helderman; T. C. Reynolds; T. B. Strom; The insulin receptor as a universal marker of activated lymphocytes. European Journal of Immunology 1978, 8, 589-595, 10.1002/eji.1830080810.
- Henrike J. Fischer; Christopher Sie; Eric Schumann; Ann-Kathrin Witte; Ralf Dressel; Jens Van Den Brandt; Holger M. Reichardt; The Insulin Receptor Plays a Critical Role in T Cell Function and Adaptive Immunity. The Journal of Immunology 2017, 198, 1910-1920, 10.4049/jimmunol.1601011.
- Katrin Singer; Wan-Chen Cheng; Marina Kreutz; Ping-Chih Ho; Peter J. Siska; Immunometabolism in cancer at a glance. Disease Models & Mechanisms 2018, 11, 276, 10.1242/dmm.034272.
- Linda V Sinclair; Julia Rolf; Elizabeth Emslie; Yun-Bo Shi; Peter M Taylor; Doreen A Cantrell; Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nature Immunology 2013, 14, 500-508, 10.1038/ni.2556.
- Luc Pilotte; Pierre Larrieu; Vincent Stroobant; Didier Colau; Eduard Dolušić; Raphaël Frédérick; Etienne De Plaen; Catherine Uyttenhove; Johan Wouters; Bernard Masereel; et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proceedings of the National Academy of Sciences 2012, 109, 2497-2502, 10.1073/pnas.1113873109.
- Tomohisa Yamamoto; Hiroaki Yanagimoto; Sohei Satoi; Hideyoshi Toyokawa; Satoshi Hirooka; So Yamaki; Rintaro Yui; Jun Yamao; Songtae Kim; A-Hon Kwon; et al. Circulating CD4+CD25+ Regulatory T Cells in Patients With Pancreatic Cancer. Pancreas 2012, 41, 409-415, 10.1097/mpa.0b013e3182373a66.
- Shou-Wang Cai; Shizhong Yang; Jie Gao; Ke Pan; Ji-Ye Chen; Yu-Lan Wang; Li-Xin Wei; Jia-Hong Dong; Prognostic significance of mast cell count following curative resection for pancreatic ductal adenocarcinoma. Surgery 2011, 149, 576-584, 10.1016/j.surg.2010.10.009.
- Anton Deicher; Roland Andersson; Bobby Tingstedt; Gert Lindell; Monika Bauden; Daniel Ansari; Targeting dendritic cells in pancreatic ductal adenocarcinoma.. Cancer Cell International 2018, 18, 85, 10.1186/s12935-018-0585-0.
- Katrin Dietl; Kathrin Renner; Katja Dettmer; Birgit Timischl; Karin Eberhart; Christoph Dorn; Claus Hellerbrand; Michael Kastenberger; Leoni Kunz-Schughart; Peter J. Oefner; et al. Lactic Acid and Acidification Inhibit TNF Secretion and Glycolysis of Human Monocytes. The Journal of Immunology 2009, 184, 1200-1209, 10.4049/jimmunol.0902584.
- Sen Yang; Qiaofei Liu; Quan Liao; Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Frontiers in Cell and Developmental Biology 2021, 8, 132, 10.3389/fcell.2020.607209.
- Evanna L. Mills; Luke A. O'neill; Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. European Journal of Immunology 2016, 46, 13-21, 10.1002/eji.201445427.
- Juan-Carlos Rodríguez-Prados; Paqui G. Través; Jimena Cuenca; Daniel Rico; Julián Aragonés; Paloma Martín-Sanz; Marta Cascante; Lisardo Boscá; Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. The Journal of Immunology 2010, 185, 605-614, 10.4049/jimmunol.0901698.
- Arvand Haschemi; Paul Kosma; Lars Gille; Charles R. Evans; Charles F. Burant; Philipp Starkl; Bernhard Knapp; Robert Haas; Johannes A. Schmid; Christoph Jandl; et al. The Sedoheptulose Kinase CARKL Directs Macrophage Polarization through Control of Glucose Metabolism. Cell Metabolism 2012, 15, 813-826, 10.1016/j.cmet.2012.04.023.
- Luke A. J. O'neill; Rigel J. Kishton; Rigel J. Kishton Jeff Rathmell; A guide to immunometabolism for immunologists. Nature Reviews Immunology 2016, 16, 553-565, 10.1038/nri.2016.70.
- Amedeo Amedei; Elena Niccolai; Domenico Prisco; Pancreatic cancer: Role of the immune system in cancer progression and vaccine-based immunotherapy. Human Vaccines & Immunotherapeutics 2014, 10, 3354-3368, 10.4161/hv.34392.
- Lucia De Monte; Michele Reni; Elena Tassi; Daniela Clavenna; Ilenia Papa; Helios Recalde; Marco Braga; Valerio Di Carlo; Claudio Doglioni; Maria Pia Protti; et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. Journal of Experimental Medicine 2011, 208, 469-478, 10.1084/jem.20101876.
- David O’Sullivan; Gerritje J.W. van der Windt; Stanley Ching-Cheng Huang; Jonathan D. Curtis; Chih-Hao Chang; Michael D. Buck; Jing Qiu; Amber M. Smith; Wing Y. Lam; Lisa M. DiPlato; et al. Memory CD8+ T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 2014, 41, 75-88, 10.1016/j.immuni.2014.06.005.
- I Esposito; M Menicagli; N Funel; F Bergmann; U Boggi; F Mosca; G Bevilacqua; D Campani; Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. Journal of Clinical Pathology 2004, 57, 630-636, 10.1136/jcp.2003.014498.
- Matthew G. Vander Heiden; Lewis C. Cantley; Craig B. Thompson; Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029-1033, 10.1126/science.1160809.
- Carina Arnold; Philipp Demuth; Nina Seiwert; Simon Wittmann; Kerstin Boengler; Birgit Rasenberger; Markus Christmann; Magdalena Huber; Thomas Brunner; Michael Linnebacher; et al. The Mitochondrial Disruptor Devimistat (CPI-613) Synergizes with Genotoxic Anticancer Drugs in Colorectal Cancer Therapy in a Bim-Dependent Manner. Molecular Cancer Therapeutics 2022, 21, 100-112, 10.1158/1535-7163.mct-21-0393.
- Rita Rebelo; Bárbara Polónia; Lúcio Santos; M. Vasconcelos; Cristina Xavier; Drug Repurposing Opportunities in Pancreatic Ductal Adenocarcinoma. Pharmaceuticals 2021, 14, 280, 10.3390/ph14030280.
- Mohammad Mazharul Islam; Andrea Goertzen; Pankaj K. Singh; Rajib Saha; Exploring the metabolic landscape of pancreatic ductal adenocarcinoma cells using genome-scale metabolic modeling. iScience 2022, 25, 632, 10.1016/j.isci.2022.104483.