Innate immune response is one of our primary defense against pathogens infection, although, if dysregulated, it represents the leading cause of chronic tissue inflammation. This dualism is even more present in the central nervous system, where neuroinflammation is both important for the activation of reparatory mechanisms and, at the same time, leads to the release of detrimental factors that induce neurons loss. Key players in modulating the neuroinflammatory response are mitochondria. They are responsible for a variety of cell mechanism that control tissue homeostasis, such as autophagy, apoptosis, energy production and also inflammation. Accordingly, it is widely recognized that mitochondria exert a pivotal role in the development of neurodegenerative diseases, since the neurodegenerative process is highly based on neuroinflammation and tissue damage. Interestingly, it has been suggested that neuroinflammation, and thus mitochondria (dys)function, have a fundamental role in neurodegenerative diseases and also in acute brain damage, such in ischemic stroke and epileptic seizures.
- Neuroinflammation
- mitochondria
- neurodegeneration
- inflammasome
- ROS
- cGAS-STING
- mitophagy
- DAMPs
- Introduction: The Cellular Players of Neuroinflammation
The new century, together with technological innovations, brought new insight into the intrinsic communication between the central nervous system (CNS) and the innate immune response. It was a common thought that the brain was a privileged tissue of our body, due to the presence of the blood–brain barrier (BBB) that would have avoided the access of immune cells [1,2]. This hypothesis has been challenged by an increasing number of studies, becoming nowadays an obsolete consideration, even though the CNS still conserves some unique immunological features [3]. Specifically, immune cells reside at the meninges granting surveillance to the brain, and meninges are provided of lymphatic vessels, able to drain large particles and immunomodulatory cytokines directly to the peripheral immune system through lymph nodes connections [4,5]. Nevertheless, the resident key players of the neuroimmune system are glial cells. These CNS immune cells are classified as macroglia (oligodendrocytes and astrocytes) and microglia, they regulate several physiological processes required for neuronal survival and brain function.
As far as we are now aware, besides being part of glial cells, oligodendrocytes do not have a major role in the physiological neuroinflammation, since they mainly provide physical and metabolic support to neurons promoting the myelinating process [6]. Noticeably, oligodendrocyte gap junctions’ deficiency due to genetic defects has been associated with increased neuroinflammation in mouse models, indicating that the altered expression of connexins in oligodendrocytes, besides being a consequence of inflammation, can also promote a proinflammatory environment [7]. Astrocytes are the most numerous glial cells of the CNS, exerting diverse roles, such as the regulation of synaptic plasticity and, more broadly, the control of brain homeostasis, also by coordinating local energy metabolism. Importantly, they also play a role in neuroprotection by maintaining the BBB intact, due to their tight interactions with the cerebrovascular endothelium [8,9]. Furthermore, astrocytes release proinflammatory cytokines, such as tumor necrosis factor α (TNF-α), which besides boosting the local inflammatory response by acting on microglia and neurons, is important in facilitating lymphocytes crossing the BBB into CNS parenchyma [10]. Accordingly, abnormal astrocytes activation, mainly characterized by hypertrophy of soma and processes, plays a key role in the neuroinflammatory process, also owed to the strict communication with microglia [11]. Being firstly described a century ago by Pio del Rio Hortega as the ‘third element’ of the CNS [12], microglia cells are now defined as the innate immune cells of the CNS characterized by the expression of CX3CR1, CD11b, Iba1, and F4/80 markers, by their myeloid origin, and by their phagocytic ability [13]. Microglia exert different functions in the CNS: they are responsible for sensing changes in the surrounding microenvironment, including both physiological changes and pathogens invasion, thus activating either their housekeeping or defense function [14].
Neuroinflammation is a natural process of defense, precisely and timely regulated, which includes a proinflammatory phase aimed to neutralize the danger, and an anti-inflammatory phase that restores the tissue homeostasis by activating the regenerative processes. While an acute neuroinflammatory response reduces injury by contributing to the repair of damaged tissue, chronic glial activation, which results from persistent stimuli, is a fundamental component of neurodegenerative diseases, and contributes to neuronal dysfunction, and therefore to CNS diseases progression [15]. As a consequence, the neuroimmune response performed by activated glial cells has a dichotomous role in the CNS. On one side, it induces the activation of repairing and regenerating mechanisms (i.e., remyelination), while on the other, the uncontrolled release of inflammatory mediators as proinflammatory cytokines, reactive oxygen species (ROS), and nitric oxide (NO) boost a chronic neuroinflammatory state, and is potentially dangerous for the neighboring cells. The aberrant release of these inflammatory molecules, together with the consequent upregulation of immune receptors on the other CNS cells, lead to tissue damage and the consequent activation of peripheral B- and T-cell responses due to the meningeal lymphatic system drainage [16]. This cascade of events enhances the inflammatory process owing to the synergistic action of microglia and lymphocytes against the antigen presenting cells [17,18].
Acute neuroinflammation usually takes place during infectious disease or during chronic autoimmune disorders such as multiple sclerosis (MS), but recent evidence has suggested how prolonged neuroinflammation is a ubiquitous pathological sign of several neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases (PD and AD) [19,20,21]. Accordingly, the close link between neuroinflammatory state and neurodegeneration suggests that neuroimmune mechanisms might trigger neuronal degeneration, resulting in neurotoxicity and neuronal cell loss [22,23]. Interestingly, the presence of mitochondrial dysfunctions both in neurodegenerative and neuroinflammatory CNS pathologies might represent the key connection between chronic immune activation and neuronal degeneration [24,25,26]. Mitochondria are organelles of endosymbiotic bacterial origin involved in various cellular functions, from the regulation of energy production and metabolism to the control of cell proliferation and programmed cell death [27]. Noticeably, mitochondria are also endowed with the ability to sense and react to cellular damage and to promote efficient host immune response by producing secondary messengers fundamental in the activation of immune cells and by contributing to the activation of inflammasomes, i.e., of the intracellular protein complexes that detect and respond to danger stimuli. Therefore, it is not surprising that increasing literature is supporting the central role of these organelles in the pathogenesis of both inflammatory and neurodegenerative CNS disorders.
- Role of Mitochondria in Neuroinflammation
Neuroinflammation is an innate inflammatory response within the CNS against harmful and toxic stimuli, mediated by the activation of resident immune cells, by the recruitment of peripheral lymphocytes and, lastly but most importantly, by the production of cytokines, chemokines, ROS, and other proinflammatory secondary messengers. The main cellular players involved in the neuroinflammatory process are glial cells, such as astrocytes and microglia. For a long time, glial cells residing in a healthy brain were defined as inactive. Following damage or infection, glial cells become “activated”, even though the terms “resting” and “activated” are vague and obsolete due to the high plasticity of these cells, which have shown to be able to dynamically shift between a spectrum of different phenotypes [28] (Figure 1). In fact, the advent of in vivo techniques, such as 2-photon microscopy, allowed the discovery that in their “resting” state, microglial cells are instead highly active, by surveying their microenvironment with extremely motile processes and protrusions [29]. Additionally, astrocytes, the most abundant glial cell population, participate in the immune and inflammatory responses of the CNS by sensing both exogenous and endogenous material through the expression of specific receptors. Indeed, even if mainly expressed by microglial cells, pattern recognition receptors (PRRs) that are fundamental for the primary recognition of infectious agents and of endogenous danger signals, are also expressed by astrocytes [30]. PRRs, localized on the cell surface, in the endosomes and also in the cytoplasm, upon the recognition of a specific antigen lead to intracellular signaling cascade ending with the release of proinflammatory mediators [31]. It is important to underline that astrocytes mainly rely on microglia for their activation. In fact, microglia control the surrounding microenvironment by using their dynamic ramifications to sense and detect any occurring alteration in brain homeostasis: once in contact with dangerous molecular factors, microglia acquire a less ramified phenotype, starting their immunomodulatory activity either by phagocytosis or by proinflammatory factors secretion [32]. Several molecular pathways are involved in activating and maintaining the inflammatory state within the CNS: PRRs signaling, cytokine receptor signaling, triggering receptor expressed on myeloid cells-2 (TREM2) signaling, ROS production, and secretion [32,33]. Interestingly, an increasing number of studies demonstrate the direct involvement of mitochondria in the modulation of the innate immune response by their participation in PRRs signaling, ROS production, and thus inflammasome assembly [34,35,36], as shown in Figure 1. PRRs are are present on inflammatory cells like macrophages, neutrophils, dendritic cells, microglia and astrocytes and responsible for the recognition of pathogens. They can be expressed both on cell membrane, as for the Toll-Like Receptors (TLR), and in the intracellular compartments as for the nucleotide-binding oligomerization domain-like receptors (NLRs) and cyclic GMP-AMP synthase (cGAS) [38]. As a part of the innate immune system, inflammation is initiated when PRRs detect pathogen-associated molecular patterns (PAMPs), such as microbial nucleic acids, lipoproteins, and carbohydrates, or DAMPs that are commonly released from injured cells following stress conditions [39,40]. For examples mitochondrial DNA (mtDNA) is a circular molecule of double‐stranded (ds)DNA, responsible for the activation of the cGAS-STING pathway upon mitochondria damage. In the same way, mtDNA, together with extracellular ATP, ROS, RNS and other mitochondria derived DAMPs, are also responsible for the activation of the NLRP3 inflammasome and the consequent release of pro-inflammatory cytokines, as IL-1b and IL-18. The excessive ROS production, besides activating the NLRP3 inflammasome, leads also to mitochondrial depolarization and therefore to mitochondrial damage and/or dysfunction. To counteract these pro-inflammatory mechanisms, mitochondria can undergo a selective autophagic process called mitophagy, responsible for the organized digestion of damaged mitochondria [82]. Due to the importance of mitochondria in regulating neuroinflammation, mitophagy represents a key factor in modulating DAMPs response, by preventing their release both in the cytoplasm and in the extracellular space, thus limiting the occurrence of sustained inflammatory events.
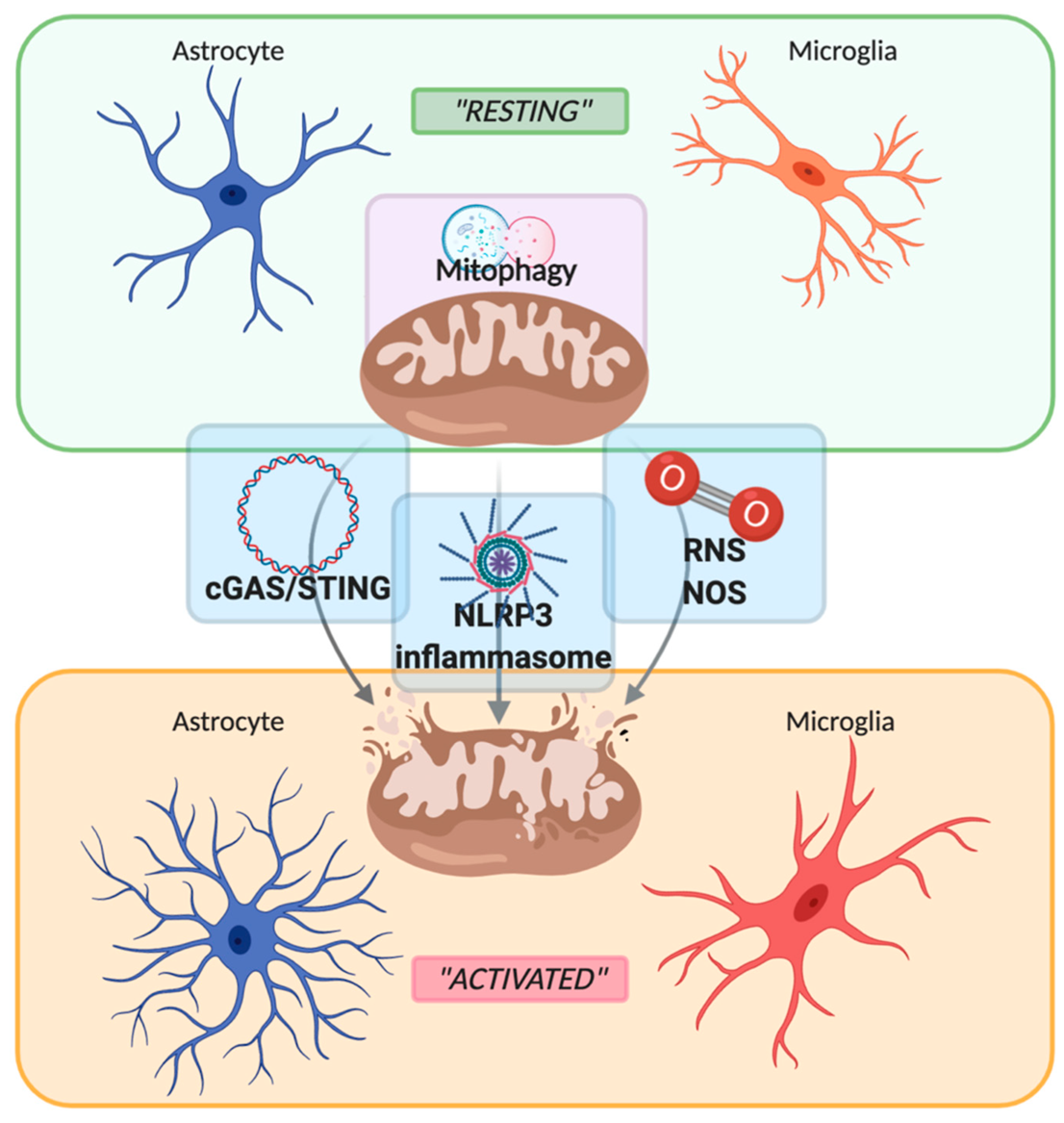
Figure 1. Schematic representation of the switch between “resting” and “activated” state of astrocyte and microglia mediated by mitochondria. Mitochondria maintain their healthy and physiological state by mitophagy. Upon stressful condition, such as inflammatory stimuli, mitochondria are disrupted with the consequent release of mtDNA, damage-associated molecular patterns (DAMPs), reactive oxygen/nitrogen species (ROS and RNS), leading to the activation of the sentinel of the central nervous system such as astrocytes and microglia. Created with BioRender.com.
- Conclusions
Neuroinflammation has been shown to play a pivotal role in CNS disorders, being mainly responsible for the neuronal cell loss and the exacerbation of the pathology. As widely described, mitochondria have a prominent part in this process: they are responsible for inflammasome assembly and ROS production, and they enclose a large amount of DAMPs, accountable for sustaining the inflammatory process. Interestingly, all these proinflammatory roles are balanced by mitophagy, which is responsible for eliminating abnormal mitochondria in order to interrupt the inflammatory sprouts. These aspects are finely balanced in CNS physiological homeostasis, since acute inflammation cover also a protective role in case of pathogens infection, but the minimal perturbation of this fine regulation triggers the neurodegenerative process. As neurodegeneration involves many altered pathways, researchers have focused the attention on many possible targets including abnormal oxidative stress and uncontrolled neuroinflammation.
Researchers are investigating why pathways that are crucial for cell survival are dysregulated in neurodegenerative diseases. For instance, neuroinflammation is vital in activating the regenerative process in IS and epilepsy, but its atypical stimulation leads to opposite results. The actual used therapies act mainly on symptomatic relief, and different pharmacological mechanisms of used drugs are yet to be fully elucidated. At the same time, even if many preclinical studies reported encouraging results, various clinical trials have shown controversial outcome or poor efficacy. This scenario could be ascribed to one of the biggest challenges of clinical trials, that is patients’ heterogeneity and the presence of comorbidities. For these reasons, as far as now, no therapies have shown efficacy in preventing neurodegenerative diseases or at least in significantly reducing their progression. However, an increasing number of studies are focusing on deregulated neuroinflammatory pathways as a common feature in NDDS. In particular, in MS, due to its autoimmune nature, drugs targeting neuroinflammation have already been approved and have displayed the most prominent results.
In conclusion, even if many aspects of inflammation-driven neurodegeneration are still unclear and further studies are needed to exploit all the pathways beyond this phenomenon, dysregulated inflammatory responses appear as a common feature for progression of brain diseases. Therefore, targeting neuroinflammation, also by acting on mitochondria, in NDDs, IS, and epilepsy represents a promising complementary therapy to obtain better clinical outcomes.
This entry is adapted from the peer-reviewed paper 10.3390/biom10101437