Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Immunotherapy is a powerful clinical strategy for the treatment of various diseases, including cancer, and an understanding of cancer immunology is important to the optimization of this strategy to achieve higher efficacy.
- cancer immunotherapy
- delivery systems
- nanoparticles
- biomaterials
1. Cancer Immunotherapy
Nearly a decade after Science named ’cancer immunotherapy’ as the breakthrough of the year 2013 [1], it has seen remarkable advances over the years. Many preclinical studies yielded novel therapies that became successful upon enrollment in clinical trials. Such success is striking in solid tumors [2]. Thus, immunotherapy is a powerful clinical strategy for the treatment of various diseases, including cancer [3], and an understanding of cancer immunology is important to the optimization of this strategy to achieve higher efficacy. For instance, advancements in single-cell RNA sequencing technologies have provided the opportunity to dissect heterogeneous tumor cells. Interrogation of this TME milieu has given clues to the precise nature of tumor-infiltrating cells and other intratumoral immune cells. Moreover, cancer immunotherapy can manipulate the immune system to identify and fight cancer cells, thereby inducing a durable response [4] with the overall aim of providing active or passive immunity against tumors [5]. Over the years, oncologists have depended on only three treatment options: surgical resection, radiotherapy, and chemotherapy. In addition, the use of small molecule inhibitors for certain kinases in many clinical procedures is still an ongoing practice in precision oncology. However, the emergence of immune-based cancer therapy has improved the choice of treatment and cancer management strategies. Since the discovery and use of the first immunotherapy, scientists have developed several other immunotherapies, including immune checkpoint inhibitors (ICIs), adoptive cell transfer, cytokines, vaccines, and others (Figure 1), which are discussed in detail below.
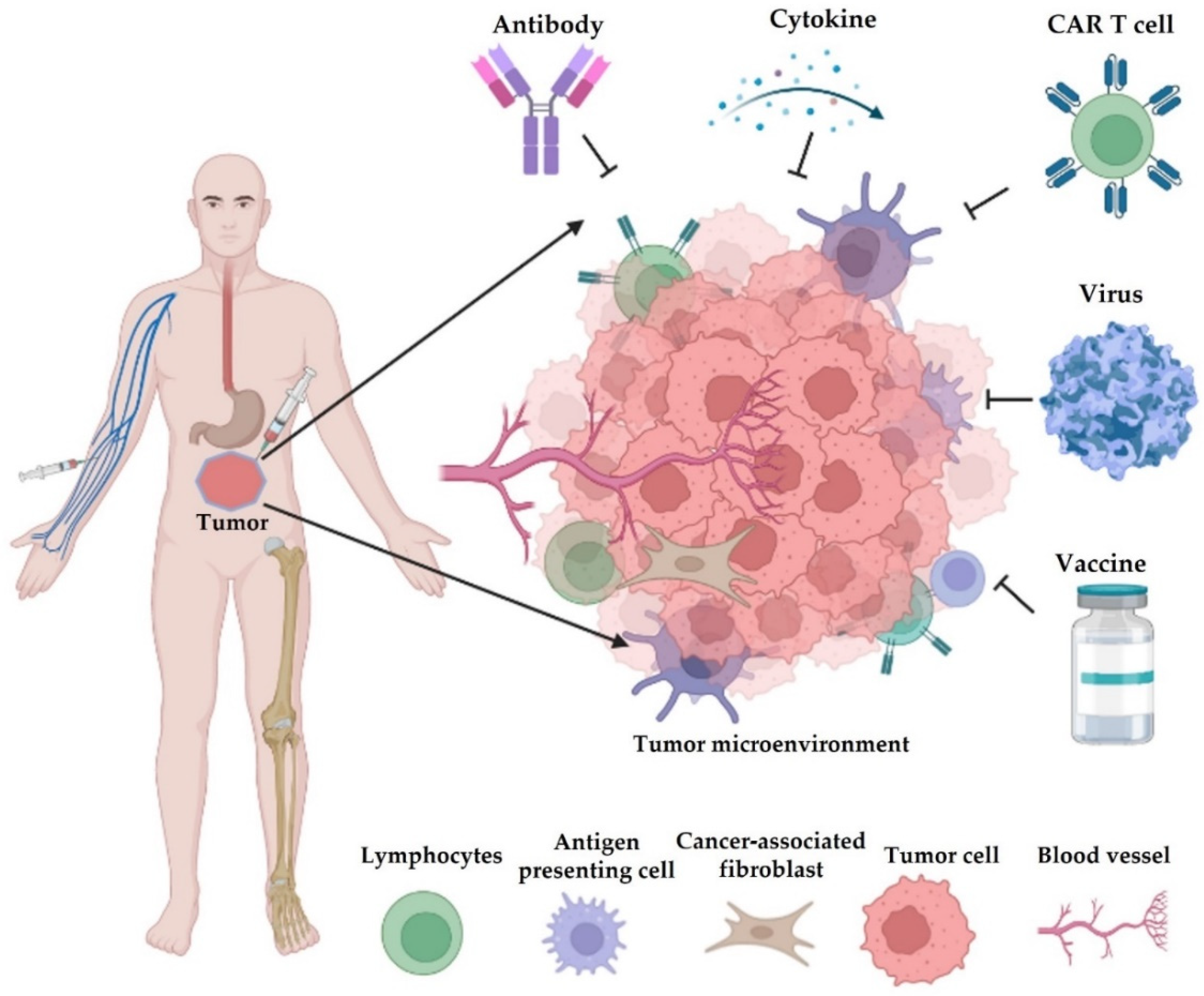
Figure 1. Different types of cancer immunotherapy. They mainly consist of blockade of immune checkpoints (e.g., antibody), adoptive transfer of engineered cells (e.g., chimeric antigen receptor (CAR) T cells, natural killer/NK cells, and macrophages), cytokine therapy, infection of oncolytic viruses, and cancer vaccines. Most of these therapeutics are administered by intravenous injection (i.v.), and some drugs are given by subcutaneous (s.c.), intraperitoneal (i.p.), or intramuscular (i.m.) injections.
1.1. Checkpoint Inhibitors
The discovery of immune checkpoints, such as CTLA-4 and PD-1, has revolutionized cancer immunotherapy [6]. These checkpoints interact with their cognate ligands on tumors and quell antitumor T cell responses. A paradigm for T cell activation includes an initial presentation of a major histocompatibility complex (MHC)-anchored antigen to T cells, interaction with the co-stimulatory receptor, and cytokine stimulation. Another inhibitory co-receptor exists to provide a negative regulation of T cell activation. These inhibitory co-receptors are checkpoint proteins that can induce adaptive tolerance and T cell exhaustion [7]. Notably, immune checkpoints regulate the tumor-killing effect of immune effector cells. Thus, ICIs can target the dysfunctional immune system to restore the effector function of cytotoxic CD8 T cells [8][9].
The first identified immune checkpoint was CTLA-4 [10]. This receptor usually out-competes another cell surface receptor, CD28, on T cells for their costimulatory ligands, CD80 and CD86. Antibodies targeted against CTLA-4 can enhance T cell response to tumors. Ipilimumab is a monoclonal antibody and the first globally approved anti-CTLA-4 for the first or second line of treatment in patients with malignant melanoma [11]. Apart from the use of ipilimumab as a single-agent monotherapy, it has been used in combination with other therapies for the treatment of various malignancies [12][13][14]. Many combination strategies that can block CTLA-4 or other immune checkpoints have been evaluated in several clinical trials around the globe. Mechanistically, anti-CTLA-4 induces preferential ligation of CD80/CD86 to CD28, leading to T cell activation.
PD-1 is another key checkpoint receptor that can modulate T cell activities in order to promote self-tolerance and activate the senescence of antigen-dependent T cells while preventing the apoptosis of regulatory T cells (Tregs). Cancer cells incessantly explore this mechanism by upregulating PD-L1, a cognate ligand of PD-1. Immunotherapy based on PD-1 blockade has shown promising efficacy in both solid and hematological malignancies [15]. In 2014, the FDA approved anti-PD-1 nivolumab, a fully humanized immunoglobulin G4 monoclonal antibody, for the treatment of advanced melanoma [15][16]. This immunotherapy can transform patient cohorts with microsatellite unstable colorectal cancer [17]. Since its initial approval, nivolumab has been repurposed and approved for the treatment of other malignancies. These include NSCLC, renal cell cancer, Hodgkin’s lymphoma, squamous head and neck cancer, urothelial carcinoma, and HCC [18][19]. More recently, the FDA approved cemiplimab (PD-1 inhibitor) for the first-line treatment of advanced non-small cell lung cancer [20], which has been approved for patients with metastatic cutaneous squamous cell carcinoma (CSCC) or patients with locally advanced CSCC that is not suitable for curative surgery or radiation [21].
Another humanized monoclonal anti-PD1 antibody is pembrolizumab. Following its previous approval for the treatment of NSCLC and unresectable melanoma [22], the U.S. FDA approved this ICI for the treatment of patients with advanced PD-L1-positive gastric and gastroesophageal junction adenocarcinoma who have progressed on at least two lines of chemotherapies [23]. A recent report has shown that pembrolizumab can be used as the first line of treatment for recurrent and metastatic HNSCC [24]. In a clinical trial, pembrolizumab showed safety and efficacy signals in phases 1 and 2 for the treatment of classic Hodgkin’s lymphoma [25].
Immunotherapies that can target the PD-1 ligand, PD-L1, have been developed. Atezolizumab is anti-PD-L1 immunotherapy, and the first ICI approved for the treatment of triple-negative breast cancer [26]. Current preclinical and clinical evidence suggests that atezolizumab may be approved as a single monotherapy or in combination with other therapies for the treatment of various malignancies. In addition to atezolizumab, two additional anti-PD-L1 immunotherapies have been approved. One, durvalumab, was approved for the treatment of urothelial cancer [27] and extensive-stage SCLC patients [28]. The other, namely avelumab, is a human IgG1 approved for the treatment of Merkell cell carcinoma and urothelial carcinoma [29].
1.2. Cytokine Therapies
Cytokines play an important role in the regulation of innate and adaptive immunity while acting as messengers via autocrine and paracrine signalings over a short distance [30]. Certain antitumor effector functions involving critical aspects of immunity require the release of cytokines or cytokine-mediated activation of antitumor immunity. Over time, scientists have developed an interest in harnessing cytokines for the treatment of cancer.
Interferon alpha (IFN-α) belongs to the family of cytokines. Like other type I IFNs, it signals through the Janus kinase 1 (JAK1) signal transducer and activator of the transcription (STAT) pathway. IFNα polarizes CD4 T cells to T helper type 1 (Th1) effector cells, upregulates MHC class I molecules, and activates caspase-dependent apoptosis in certain cancers. For decades, various formulations of recombinant IFN-α were approved for the treatment of various malignancies, including metastatic renal cell carcinoma, acquired immunodeficiency syndrome (AIDs)-related Kaposi’s sarcoma, follicular lymphoma, chronic myelogenous leukemia, cervical intraperitoneal neoplasms, and completely resected stage III or IV high-risk melanoma [30].
Another universally approved cytokine therapy is interleukin (IL)-2, which is mainly secreted by Th1 effector cells. CD8 T cells and NK cells also secrete IL-2 but to a lesser extent [31]. While acting as a T cell growth factor, IL-2 promotes the expansion of T cells, which is important in the regulation of T cell response and the maintenance of self-tolerance via activation-induced cell death (AICD). IL-2 has been approved for the treatment of metastatic melanoma and metastatic renal cell carcinoma (mRCC). In addition, IL-2 is widely combined with adoptive T cell therapy, as it enhances the ex vivo expansion of T cells [32][33].
Other cytokines, including IFN-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-12, IL-15, and IL-21, have been evaluated for their anticancer potential in preclinical and clinical models. IFN-γ showed an initial promising result in a phase 2 trial but was not approved to treat cancer patients due to the lack of efficacy [34]. In several trials, GM-CSF demonstrated inconsistent efficacy in addition to its scarring effect [35]. With a previous promising phase 1 trial result, IL-12 belied the previous dosing regimen and showed adverse effects and mortality [36]. Further, the antitumor efficacy of IL-15 was evaluated in preclinical studies and phase 1/2 clinical trials. IL-15 activated and caused the expansion of NK, NKT, and (m) CD8 T cells [37]. In another study, the combination of IL-15 with anti-PD-L1 and anti-CTLA4 contributed to favorable OS [38]. Thus, the outcomes from these studies are promising for the future development of IL-15–based therapy. On the other hand, IL-21 plays a critical role in chronic inflammatory bowel disease (IBD) and inflammation-induced colon cancer, thus leading to its termination in the clinical trial [39]. Despite these treatment-related adverse events (TRAEs), there are several ongoing clinical trials to re-evaluate the anti-tumor potentials of these cytokines.
1.3. Adoptive Cell Transfer Therapy
Adoptive cell therapy (ACT) has emerged as an important therapy for cancers, especially personalized cancer therapy [40]. T cells for ACT mainly include tumor-infiltrating lymphocyte-derived T cells or genetically engineered T cells with the expression of conventional T cell receptors or chimeric antigen receptors [41]. Commonly used gene-editing strategies in T cells include retroviral or lentiviral transduction, zinc finger, or transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeat (CRISPR)-associated 9 (Cas9) endonuclease technology [42][43][44].
Chimeric antigen receptor T (CAR-T) cells use a gene transfer technology that involves an ex vivo modification of T cells and an adoptive transfer of the engineered T cells in order to target tumor-associated antigen (TAA) and bolster the antitumor function of T lymphocytes. Although different types of T cells may present different efficacy with a specific CAR technology, various modifications are available to prolong survival and redirect the specificity and function of T cells [45].
In general, CAR-T cells are structurally engineered to contain an extracellular antigen-binding domain derived from a single-chain variable fragment (ScFv) of a monoclonal antibody, an extracellular region containing a spacer domain, a transmembrane domain, and an intracellular domain [46]. Upon antigen binding to the ScFv, the intracellular domain is capable of initiating signaling that culminates in T cell activation. This T cell activation is MHC-independent and can lead to tumor destruction. The success of CAR-T-based immunotherapy depends on the TAA selected for CAR specificity. Usually, the CAR gene is designed to recognize only TAAs that are critical to the survival of the tumor. Despite the promising outcomes of CAR technology, certain tumor genetic mutations and epigenetic alterations are drivers of immunoediting, which can result in therapeutic resistance [47][48].
CAR-T cell-based therapy has shown encouraging efficacies toward hematological malignancies. One major TAA target is CD-19. Kymriah and Yescarta are CD19-targeting CAR-T cell products approved by the U.S. FDA for the standard of care of B cell acute lymphoblastic leukemia (B-ALL) and diffuse large B-cell lymphoma (DLBCL), respectively [49][50]. Brexucabtagene autoleucel (brexu-cel) was also approved for the treatment of relapsed or refractory (r/r) mantle cell lymphoma [51]. Other CD-19-directed CAR-T cell products include tisagenlecleucel (tisa-cel) and axicabtagene ciloleucel (axi-cel), which were approved for the treatment of patients with r/r DLBCL, B-ALL, and primary mediastinal B-cell lymphoma (PMBCL) [52][53]. In other tumor types, CAR may be engineered to recognize other antigens. For instance, ganglioside GD2 is a TAA in neuroblastoma [54]; CD70 is a novel target in gliomas [55]; CD20 and CD22 are TAAs in relapsed refractory Burkitt lymphoma [56]. In the previous study, researchers reported liver-intestine cadherin (CDH17) as a novel target in pancreatic cancer. The data showed that knockout of CDH17 suppressed Panc02-H7 growth and caused tumor regression in the orthotopic mouse model [57]. As the evolution of CAR technology continues, CDH17 has become a novel TAA for the development of newly engineered CAR T cells. Feng et al., in a 2022 report, demonstrated that CDH17CAR T cells suppressed neuroendocrine and gastrointestinal tumors without TRAEs [58].
1.4. Oncolytic Virotherapy
The use of oncolytic viruses in the treatment of malignancies is becoming increasingly promising. Oncolytic viruses are genetically modified viruses that can selectively replicate and target cancer cells for destruction without any deleterious effects on normal tissue [59]. Mechanistically, virotherapy induces specific antitumor immunity in the context of tumor-specific viral replication.
The first oncolytic virotherapy, rigvir, was developed from the native ECHO-7 strain of picornavirus. Rigvir was registered in Latvia, Georgia, Armenia, and Uzbekistan, where it was approved for the treatment of melanoma [60]. Furthermore, a genetically engineered adenovirus, oncorine (H101), was approved in November 2005 by the Chinese SFDA as a standard of care for nasopharyngeal carcinoma, but in combination with chemotherapy [61]. Other oncolytic adenovirus therapies that can target and eliminate cancer cells in both preclinical and clinical models have been developed [62][63][64]. However, the efficacy of this therapy is generally challenged by inefficient systemic delivery to the target tissue. This is due to the presence of pre-existing neutralizing antibodies to adenovirus [65] and non-specific uptake [66]. In 2015, a herpes simplex virus type 1 (HSV-1)-based virotherapy was developed. This modified HSV-1, called Talimogene laherparepvec (T-vec), was armed with GM-CSF. Following a phase 3 clinical trial that showed that T-vec significantly caused tumor regression and prolonged the OS of melanoma patients [67], the U.S. FDA approved T-vec for the treatment of unresectable cutaneous, subcutaneous, and nodal lesions in melanoma patients with relapse [59][68].
In addition to the approved virotherapies, at least 40 oncolytic viruses are currently being tested against different cancers [69]. Some of these oncolytic viral therapies have shown promising results in phase 3 clinical trials and are now awaiting approval. These include oncolytic vaccinia virus-derived pexastimogen devbacirepvec (pexa-vec), oncolytic adenovirus-derived CG0070, and oncolytic reovirus-derived reolysin (pelareorep) [70].
1.5. Cancer Vaccines
Therapeutic cancer vaccines aim to promote tumor regression, establish robust antitumor memory, and avoid adverse events [71]. In principle, cancer vaccines stemmed from the natural phenomenon of antitumor immunity that emerged from natural or chemotherapy-induced immunogenic cell death (ICD). Therapeutic cancer vaccines can be used to treat advanced or relapsed tumors that are refractory to conventional therapies [72]. During ICDs, tumor antigens are released, captured, and cross-presented by APCs, leading to their maturation and migration to secondary lymphoid organs, where they educate naïve T cells. Upon activation, T cells roll back to the TME and cause the direct destruction of cancer cells [73][74].
Cancer vaccines usually contain specific tumor antigens and are exogenously administered to activate APCs such as DCs, leading to the stimulation of an adaptive immune response against tumors containing this antigen and the resurgence of robust tumor control. Usually, large amounts of qualitative antigens are delivered to the DCs to induce optimal DC activation, culminating in sustained T cell activation, TME infiltration, and response maintenance [71]. Alternatively, it is possible to develop a cancer vaccine from an endogenous source, a method called the in situ (ISV) approach. It involves antigen sourcing from dying or dead cells in the TME [75].
Neoantigens derived from tumor mutations have been recognized as ideal targets of T cell-based immunotherapy and therapeutic cancer vaccines [76][77]. Neoantigen-targeted vaccines mainly include synthetic long peptides, nucleic acids, and cell-based vaccines [78]. Currently, many clinical trials have evaluated their safety and efficacy in patients [79][80]. Examples of some vaccines are discussed in the section on clinical trials.
Sipuleucel-T is an antigen-specific active immunotherapy agent that sensitizes the adaptive immune system [81] by activating the anti-PAP (prostatic acid phosphatase) immune response, leading to the destruction of cancer cells [82]. The U.S. FDA approved Sipuleucel-T after a double-blind, placebo-controlled, multicenter phase 3 trial in which Sipuleucel-T reduced the risk of death among patients with metastatic castration-resistant prostate cancer (mCRPC) [83]. Although Sipuleucel-T remains the only cancer vaccine approved, other cancer vaccines are currently being investigated. For instance, four cancer vaccines have been tested in phase 3 clinical trials of mCRPC patients. These include prostate cancer vaccine GVAX (a GM-CSF gene vaccine), anti-prostate-specific antigen (PSA) vaccine PROSTVAC, personalized peptide vaccination (PPV), and DC-based vaccine PCVAC/PCa [82].
In summary, immunotherapy has provided a powerful tool for cancer therapy, either alone or as a synergistic treatment. Some examples of FDA-approved treatments are listed in Table 1.
Table 1. Some examples of FDA-approved immunotherapies.
| S/N | Therapy | Type | Target | Indication | References |
|---|---|---|---|---|---|
| 1 | Ipilimumab | ICI | CTLA-4 blockade | Malignant melanoma | [11] |
| 2 | Cemiplimab | ICI | PD-1 blockade | Advanced NSCLC, metastatic CSCC | [20][21] |
| 3 | Nivolumab | ICI | PD-1 blockade | Advanced melanoma, metastatic colorectal cancer, NSCLC, renal cell cancer, Hodgkin’s lymphoma, squamous head and neck cancer, urothelial carcinoma, HCC | [17][18] |
| 4 | Pembrolizumab | ICI | PD-1 blockade | NSCLC, advanced melanoma, colorectal cancer, gastric and gastroesophageal cancer, classic Hodgkin’s lymphoma, metastatic HNSCC | [22][23][24][25][84] |
| 5 | Atezolizumab | ICI | PD-L1 blockade | Triple-negative breast cancer | [26] |
| 6 | Durvalumab | ICI | PD-L1 blockade | Urothelial cancer, ES-SCLC | [27][28] |
| 7 | Avelumab | ICI | PD-L1 blockade, ADCC | Merkell cell carcinoma, urothelial carcinoma | [29] |
| 8 | IFN-α | Cytokine therapy | Multiple mechanisms | mRCC, AIDs-related Kaposi’s sarcoma, follicular lymphoma, chronic myelogenous leukemia, cervical intraperitoneal neoplasms, and advanced melanoma | [7] |
| 9 | IL-2 | Cytokine therapy | AICD | mRCC | [9][10] |
| 10 | Kymriah | ACT | Anti-CD19 | B-ALL | [49][50] |
| 11 | Yescarta | ACT | Anti-CD19 | DLBCL | [49][50] |
| 12 | Brexucabtagene autoleucel | ACT | Anti-CD19 | R/r mantle cell lymphoma | [20] |
| 13 | Tisagenlecleucel | ACT | Anti-CD19 | DLBCL, B-ALL, and PMBCL | [21][22] |
| 14 | Axicabtagene Ciloleucel | ACT | Anti-CD19 | DLBCL, B-ALL, and PMBCL | [21][22] |
| 15 | Rigvir | OV | Tumor lysis | Melanoma | [60] |
| 16 | Oncorine (H101) | OV | Tumor lysis | Nasopharyngeal carcinoma | [61] |
| 17 | Talimogene laherparepvec (T-vec) | OV | Tumor lysis | Melanoma patients | [67] |
| 18 | Sipuleucel-T | Cancer vaccine | Activate antitumor immunity | mCRPC | [83] |
Abbreviations: ACT: adoptive cell transfer; ADCC: Ab-dependent cell cytotoxicity; B-ALL: B cell acute lymphoblastic leukemia; CSCC: cutaneous squamous cell carcinoma; DLBCL: diffuse large B-cell lymphoma; ES-SCLC: extensive-stage small cell lung cancer; HNSCC: metastatic head and neck squamous cell carcinoma; HCC: hepatocellular carcinoma; ICI: immune checkpoint inhibitor; mCRPC: metastatic castration-resistant prostate cancer; mRCC: metastatic renal cell carcinoma; NSCLC: non-small cell lung cancer; OV: oncolytic virus; PMBCL: primary mediastinal B-cell lymphoma.
2. Delivery Systems for Immunotherapy
Given that immunotherapy is an important strategy for cancer treatment with various advantages, such as preventing cancer metastasis and recurrence, the above-mentioned limitations, including inefficient delivery system, low efficacy, tumor penetration, optimization of synergistic treatment, off-target effects, and high toxicity of immunotherapeutic agents, can be resolved by delivery systems [85][86][87][88]. In addition, hypoxia, low nutrients in the TME, and the heterogeneity of tumor cells due to mutation significantly inhibit the function of immune cells [89][90]. The delivery system is also a very useful tool for effectively developing a combined therapeutic strategy. For example, therapeutic NPs can be applied to co-deliver chemo-immunotherapy combinations (e.g., doxorubicin and IL-12) to induce efficient intratumor delivery [91].
In this section, researchers describe some delivery approaches to overcome these limitations and improve cancer immunotherapy, including NP-based delivery, extracellular vesicles, implantable scaffolds, antigen-mediated delivery, and cell-based delivery.
2.1. Nanoparticle-Based Delivery
NPs can deliver antibodies or their fragments, peptides, proteins, and small molecules and their antagonists, such as IL-2, TGF-β inhibitors, CpG oligodeoxynucleotides, and anti-PD-1 mAbs [92][93]. There are many platforms for NPs, including liposomes, inorganic nanocarriers, dendrimers, polymeric systems, nucleic acid nanotechnology, and exosomes [94]. Some examples are shown in graphic cartoons (Figure 2).
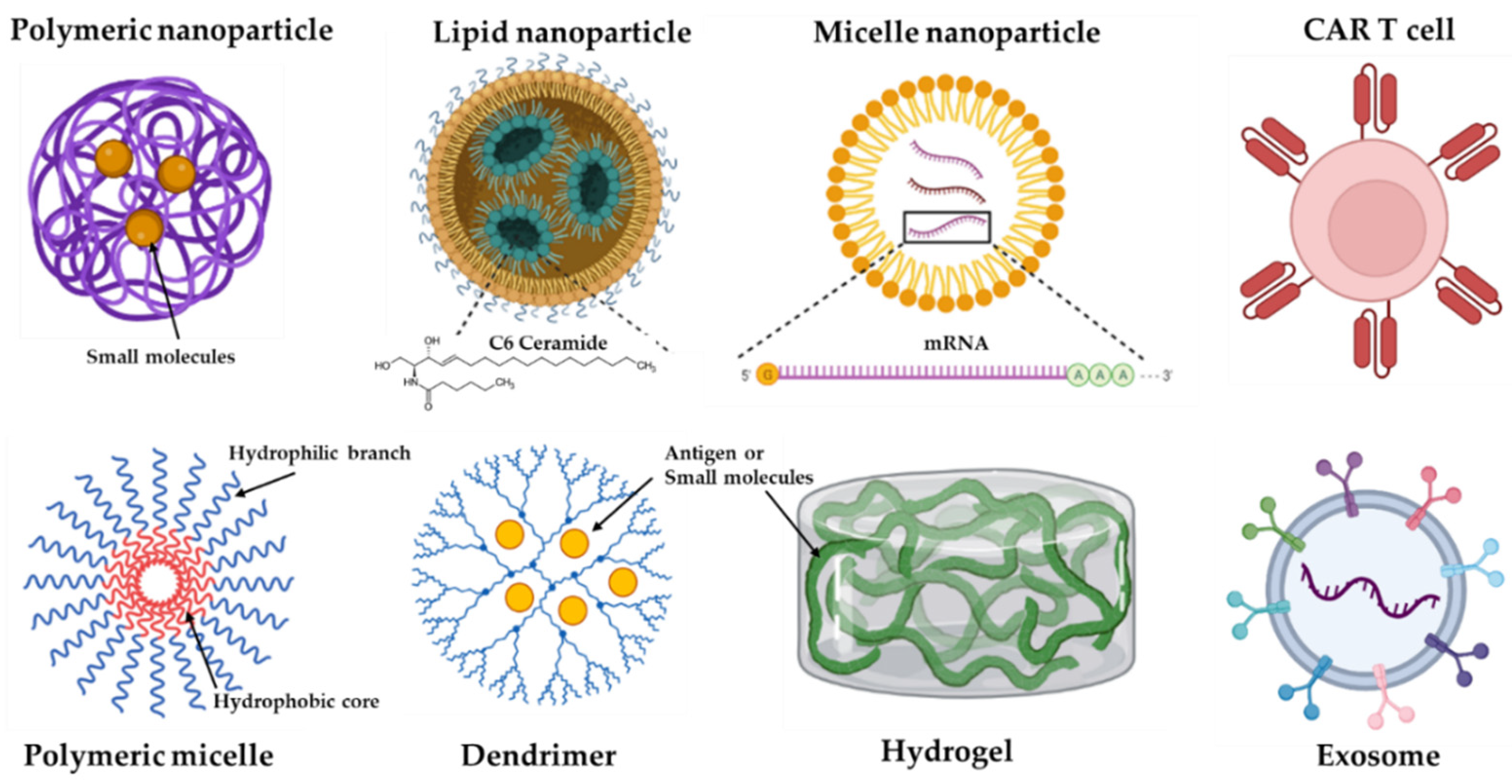
Figure 2. Some representative formats of immunotherapy delivery systems. Nanoparticles (NPs) can be formed by different materials, including iron (e.g., gold), lipid, polymeric, and self-formatting NPs.
Delivering cancer immunotherapies by NPs can increase anti-tumor efficacy, enhance drug retention, improve drug penetration, and enhance the synergetic effect of treatments [87][95][96]. For example, the self-assembling protein nanocarrier T22-GFP-H6 can selectively deliver cytotoxic agents into CXCR4-expressing tumors in an HNSCC model [97]. Furthermore, the use of NPs overcomes chemotherapeutic resistance by strategies such as inhibition of drug efflux pumps and simultaneous delivery of multiple drugs [98].
2.2. Extracellular Vesicles
Extracellular vesicles (EVs) are lipid membrane-enclosed vesicles with nanometer sizes, which are secreted by most living cells, and contain different proteins, lipids, and nucleic acid species of the source cells [99]. These EVs are mediators for the interaction of cells in the TME, regulating anti-tumor immune responses [100]. Given their delivery function, EVs have been explored as carriers of bioactive components of cancer immunotherapy. For example, EVs from fibroblast-like mesenchymal cells can be engineered to deliver siRNAs or short hairpin RNAs (shRNAs) that target oncogenic Kras to enhance anti-pancreatic cancer ability and increase mouse overall survival rates [101].
EVs can be classified into three subtypes, exosomes (30–150 nm), macrovesicles (0.1–1 μm), and apoptotic bodies (1–5 μm) based on their biogenesis mechanism [102]. For example, fibroblast activation protein-α (FAP) gene-engineered tumor cell-derived exosome-like vesicle vaccines (eNVs-FAP) can activate the maturation of DCs, elicit specific cytotoxic T cell infiltration and activation, and promote tumor ferroptosis and depletion of FAP-positive cancer-associated fibroblasts [103]. EVs have many multiple advantages as a delivery platform, including their ability to overcome natural barriers, intrinsic cell targeting properties, and circulation stability [104].
2.3. Implantable and Injectable Scaffolds
Conventionally, small drug molecules are dissolved in hydrogel for delivery, which causes drug retention with poor intratumoral delivery. One study applied a nanocomposite hydrogel (~6 nm) to deliver oxaliplatin (OXA) to treat a breast cancer cell line 4T1-induced tumor model. The results showed that this nanocomposite hydrogel significantly decreased tumor growth and metastasis by enhancing the retention and penetration of anti-cancer drugs in the TME, which also showed a synergetic effect with αPD-1 antibody [105]. Another study developed an injectable, polymerized phenylboronic acid-based immunogel for the delivery of mannan, a natural polysaccharide with the function of adjuvanticity and tumor antigen [106]. This immunogel improved anti-cancer activity against a breast cancer cell line 4 T1 cells in a mouse tumor model. Loading NPs in an injectable hydrogel formulation can yield sustained immune stimulation to inhibit cancer cell growth compared to an immediate regular I.V. or I.P. injection [107]. The inverse opal (IOPAL) 3D hydrogels have been engineered with poly(ethylene) glycol (PEG) covalently combined with heparin to resemble the lymph node microenvironment and maintain the phenotype of adoptively transferred T cells [108]. Hydrogel-mediated in situ delivery can provide many advantages, including easy use, increased local treatment agents, and prolonged treatment retention time to prevent the refraction of tumors [109].
2.4. Antigen-Mediated Delivery
The self-assembled polysaccharide nanogels of cholesteryl group-modified pullulan (CHP) can be used as antigen delivery systems for cancer immunotherapy by regulating tumor-associated macrophages (TAMs) [110]. New York esophageal squamous cell carcinoma 1 (NY-ESO-1), a cancer-testis antigen, is expressed by many cancers [111]. CHP has been applied to deliver the cancer antigen NY-ESO-1 for cancer vaccines. Patients with advanced or metastatic esophageal cancer were vaccinated with 100 μg or 200 μg of CHP-NY-ESO-1 and showed no adverse events or immunogenicity. The survival of cancer patients increased with a high dose of CHP-NY-ESO-1 treatment compared to low dose administration [112]. This strategy can provide targeted delivery and enhance the immune response [113].
2.5. Cell-Based Delivery
T cell transfer therapy, or adoptive cell transfer (ACT) therapy, is a major type of cell-based therapy, including tumor-infiltrating lymphocytes (TIL) therapy and CAR-T cell therapy [114][115]. CAR-T cell therapy is an effective and powerful immunotherapy for combating blood cancers and refractory cancers [116][117]. Unfortunately, it is also very expensive to manufacture CAR T cells [118]. State-of-the-art technology shows that delivery vectors, including lentiviruses, adenovirus-associated vectors, and nanocarriers or NPs, are commonly used for in vivo CAR (encoding nucleic acids) delivery to T cells. Receptor targeting of delivery vectors can reduce off-target cell delivery and potential toxicities [119].
In addition to the above-mentioned delivery systems, drugs themselves can form nanoscale medicines without carriers that have been designed for cancer treatment. Cargo-free nanomedicines can be classified into drug nanocrystals, prodrug self-assembled NPs, drug–drug conjugate NPS, and antibody–drug conjugates (ADCs) [120].
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics14081630
References
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433.
- van den Bulk, J.; Verdegaal, E.M.; de Miranda, N.F. Cancer immunotherapy: Broadening the scope of targetable tumours. Open Biol. 2018, 8, 180037.
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196.
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA A Cancer J. Clin. 2020, 70, 86–104.
- Baxevanis, C.N.; Perez, S.A.; Papamichail, M. Cancer immunotherapy. Crit. Rev. Clin. Lab. Sci. 2009, 46, 167–189.
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355.
- Olaoba, O.T.; Ligali, F.C.; Alabi, Z.O.; Akinyemi, A.O.; Ayinde, K.S. Of immune checkpoint maladies and remedies: The throwing of jabs in the oncogenic ring of PDAC. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188483.
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014.
- Li, H.B.; Yang, Z.H.; Guo, Q.Q. Immune checkpoint inhibition for pancreatic ductal adenocarcinoma: Limitations and prospects: A systematic review. Cell Commun. Signal. CCS 2021, 19, 117.
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1233–1238.
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab: First global approval. Drugs 2011, 71, 1093–1104.
- Paz-Ares, L.G.; Ramalingam, S.S.; Ciuleanu, T.E.; Lee, J.S.; Urban, L.; Caro, R.B.; Park, K.; Sakai, H.; Ohe, Y.; Nishio, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced NSCLC: 4-Year Outcomes from the Randomized, Open-Label, Phase 3 CheckMate 227 Part 1 Trial. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2022, 17, 289–308.
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 773–779.
- Tsang, J.; Wong, J.S.L.; Kwok, G.G.W.; Li, B.C.W.; Leung, R.; Chiu, J.; Cheung, T.T.; Yau, T. Nivolumab + Ipilimumab for patients with hepatocellular carcinoma previously treated with Sorafenib. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 589–598.
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. PD-1/PD-L1 blockade in cancer treatment: Perspectives and issues. Int. J. Clin. Oncol. 2016, 21, 462–473.
- Olaoba, O.T.; Kadasah, S.; Vetter, S.W.; Leclerc, E. RAGE Signaling in Melanoma Tumors. Int. J. Mol. Sci. 2020, 21, 8989.
- Smith, K.M.; Desai, J. Nivolumab for the treatment of colorectal cancer. Expert Rev. Anticancer Ther. 2018, 18, 611–618.
- Finkelmeier, F.; Waidmann, O.; Trojan, J. Nivolumab for the treatment of hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2018, 18, 1169–1175.
- Zhang, C.; Yang, M. The Emerging Factors and Treatment Options for NAFLD-Related Hepatocellular Carcinoma. Cancers 2021, 13, 3740.
- Sezer, A.; Kilickap, S.; Gümüş, M.; Bondarenko, I.; Özgüroğlu, M.; Gogishvili, M.; Turk, H.M.; Cicin, I.; Bentsion, D.; Gladkov, O.; et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: A multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet 2021, 397, 592–604.
- Migden, M.R.; Chandra, S.; Rabinowits, G.; Chen, C.I.; Desai, J.; Seluzhytsky, A.; Sasane, M.; Campanelli, B.; Chen, Z.; Freeman, M.L.; et al. CASE (CemiplimAb-rwlc Survivorship and Epidemiology) study in advanced cutaneous squamous cell carcinoma. Future Oncol. 2020, 16, 11–19.
- Kwok, G.; Yau, T.C.; Chiu, J.W.; Tse, E.; Kwong, Y.L. Pembrolizumab (Keytruda). Hum. Vaccines Immunother. 2016, 12, 2777–2789.
- Joshi, S.S.; Maron, S.B.; Catenacci, D.V. Pembrolizumab for treatment of advanced gastric and gastroesophageal junction adenocarcinoma. Future Oncol. 2018, 14, 417–430.
- de Sousa, L.G.; Ferrarotto, R. Pembrolizumab in the first-line treatment of advanced head and neck cancer. Expert Rev. Anticancer Ther. 2021, 21, 1321–1331.
- Al Hadidi, S.A.; Lee, H.J. Pembrolizumab for the treatment of Hodgkin Lymphoma. Expert Opin. Biol. Ther. 2020, 20, 1275–1282.
- Reddy, S.M.; Carroll, E.; Nanda, R. Atezolizumab for the treatment of breast cancer. Expert Rev. Anticancer Ther. 2020, 20, 151–158.
- Alvarez-Argote, J.; Dasanu, C.A. Durvalumab in cancer medicine: A comprehensive review. Expert Opin. Biol. Ther. 2019, 19, 927–935.
- Al-Salama, Z.T. Durvalumab: A Review in Extensive-Stage SCLC. Target. Oncol. 2021, 16, 857–864.
- Roviello, G.; D’Angelo, A.; Generali, D.; Pittacolo, M.; Ganzinelli, M.; Iezzi, G.; Manzini, N.; Sobhani, N. Avelumab in gastric cancer. Immunotherapy 2019, 11, 759–768.
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2019, 39, 6–21.
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190.
- Andersen, R.; Donia, M.; Westergaard, M.C.; Pedersen, M.; Hansen, M.; Svane, I.M. Tumor infiltrating lymphocyte therapy for ovarian cancer and renal cell carcinoma. Hum. Vaccines Immunother. 2015, 11, 2790–2795.
- Lu, Y.C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients with Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3322–3329.
- Sugaya, M.; Tokura, Y.; Hamada, T.; Tsuboi, R.; Moroi, Y.; Nakahara, T.; Amano, M.; Ishida, S.; Watanabe, D.; Tani, M.; et al. Phase II study of i.v. interferon-gamma in Japanese patients with mycosis fungoides. J. Dermatol. 2014, 41, 50–56.
- Kaufman, H.L.; Ruby, C.E.; Hughes, T.; Slingluff, C.L., Jr. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J. Immunother. Cancer 2014, 2, 11.
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. CII 2014, 63, 419–435.
- Zhang, S.; Zhao, J.; Bai, X.; Handley, M.; Shan, F. Biological effects of IL-15 on immune cells and its potential for the treatment of cancer. Int. Immunopharmacol. 2021, 91, 107318.
- Waldmann, T.A.; Dubois, S.; Miljkovic, M.D.; Conlon, K.C. IL-15 in the Combination Immunotherapy of Cancer. Front. Immunol. 2020, 11, 868.
- Steele, N.; Anthony, A.; Saunders, M.; Esmarck, B.; Ehrnrooth, E.; Kristjansen, P.E.; Nihlén, A.; Hansen, L.T.; Cassidy, J. A phase 1 trial of recombinant human IL-21 in combination with cetuximab in patients with metastatic colorectal cancer. Br. J. Cancer 2012, 106, 793–798.
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68.
- Met, Ö.; Jensen, K.M.; Chamberlain, C.A.; Donia, M.; Svane, I.M. Principles of adoptive T cell therapy in cancer. Semin. Immunopathol. 2019, 41, 49–58.
- Singh, N.; Shi, J.; June, C.H.; Ruella, M. Genome-Editing Technologies in Adoptive T Cell Immunotherapy for Cancer. Curr. Hematol. Malig. Rep. 2017, 12, 522–529.
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815.
- Berdien, B.; Mock, U.; Atanackovic, D.; Fehse, B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014, 21, 539–548.
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric antigen receptors for T cell immunotherapy: Current understanding and future directions. J. Gene Med. 2012, 14, 405–415.
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53.
- Goldberger, O.; Volovitz, I.; Machlenkin, A.; Vadai, E.; Tzehoval, E.; Eisenbach, L. Exuberated numbers of tumor-specific T cells result in tumor escape. Cancer Res. 2008, 68, 3450–3457.
- Dai, E.; Zhu, Z.; Wahed, S.; Qu, Z.; Storkus, W.J.; Guo, Z.S. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 2021, 20, 171.
- Fournier, C.; Martin, F.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Apetoh, L. Trial Watch: Adoptively transferred cells for anticancer immunotherapy. Oncoimmunology 2017, 6, e1363139.
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560.
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342.
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544.
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380.
- Jin, L.; Ge, H.; Long, Y.; Yang, C.; Chang, Y.E.; Mu, L.; Sayour, E.J.; De Leon, G.; Wang, Q.J.; Yang, J.C.; et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro-Oncology 2018, 20, 55–65.
- Du, J.; Zhang, Y. Sequential anti-CD19, 22, and 20 autologous chimeric antigen receptor T-cell (CAR-T) treatments of a child with relapsed refractory Burkitt lymphoma: A case report and literature review. J. Cancer Res. Clin. Oncol. 2020, 146, 1575–1582.
- Liu, X.; Huang, Y.; Yuan, H.; Qi, X.; Manjunath, Y.; Avella, D.; Kaifi, J.T.; Miao, Y.; Li, M.; Jiang, K.; et al. Disruption of oncogenic liver-intestine cadherin (CDH17) drives apoptotic pancreatic cancer death. Cancer Lett. 2019, 454, 204–214.
- Feng, Z.; He, X.; Zhang, X.; Wu, Y.; Xing, B.; Knowles, A.; Shan, Q.; Miller, S.; Hojnacki, T.; Ma, J.; et al. Potent suppression of neuroendocrine tumors and gastrointestinal cancers by CDH17CAR T cells without toxicity to normal tissues. Nat. Cancer 2022, 3, 581–594.
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379.
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur. J. Pharmacol. 2018, 837, 117–126.
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr. Cancer Drug Targets 2018, 18, 171–176.
- Bazan-Peregrino, M.; Garcia-Carbonero, R.; Laquente, B.; Álvarez, R.; Mato-Berciano, A.; Gimenez-Alejandre, M.; Morgado, S.; Rodríguez-García, A.; Maliandi, M.V.; Riesco, M.C.; et al. VCN-01 disrupts pancreatic cancer stroma and exerts antitumor effects. J. Immunother. Cancer 2021, 9, e003254.
- Jung, K.H.; Choi, I.K.; Lee, H.S.; Yan, H.H.; Son, M.K.; Ahn, H.M.; Hong, J.; Yun, C.O.; Hong, S.S. Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett. 2017, 396, 155–166.
- Man, Y.K.S.; Davies, J.A.; Coughlan, L.; Pantelidou, C.; Blázquez-Moreno, A.; Marshall, J.F.; Parker, A.L.; Halldén, G. The Novel Oncolytic Adenoviral Mutant Ad5-3Δ-A20T Retargeted to αvβ6 Integrins Efficiently Eliminates Pancreatic Cancer Cells. Mol. Cancer Ther. 2018, 17, 575–587.
- Mast, T.C.; Kierstead, L.; Gupta, S.B.; Nikas, A.A.; Kallas, E.G.; Novitsky, V.; Mbewe, B.; Pitisuttithum, P.; Schechter, M.; Vardas, E.; et al. International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: Correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine 2010, 28, 950–957.
- Khare, R.; May, S.M.; Vetrini, F.; Weaver, E.A.; Palmer, D.; Rosewell, A.; Grove, N.; Ng, P.; Barry, M.A. Generation of a Kupffer cell-evading adenovirus for systemic and liver-directed gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1254–1262.
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2780–2788.
- Mondal, M.; Guo, J.; He, P.; Zhou, D. Recent advances of oncolytic virus in cancer therapy. Hum. Vaccines Immunother. 2020, 16, 2389–2402.
- Marchini, A.; Ilkow, C.S.; Melcher, A. Oncolytic Virus Immunotherapy. Cancers 2021, 13, 3672.
- Taguchi, S.; Fukuhara, H.; Todo, T. Oncolytic virus therapy in Japan: Progress in clinical trials and future perspectives. Jpn. J. Clin. Oncol. 2019, 49, 201–209.
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378.
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475.
- Alloatti, A.; Kotsias, F.; Magalhaes, J.G.; Amigorena, S. Dendritic cell maturation and cross-presentation: Timing matters! Immunol. Rev. 2016, 272, 97–108.
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013.
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148.
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356.
- Holm, J.S.; Funt, S.A.; Borch, A.; Munk, K.K.; Bjerregaard, A.-M.; Reading, J.L.; Maher, C.; Regazzi, A.; Wong, P.; Al-Ahmadie, H.; et al. Neoantigen-specific CD8 T cell responses in the peripheral blood following PD-L1 blockade might predict therapy outcome in metastatic urothelial carcinoma. Nat. Commun. 2022, 13, 1935.
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128.
- Song, Q.; Yang, B.; Sheng, W.; Zhou, Z.; Zhang, T.; Qin, B.; Ji, L.; Li, P.; Wang, D.; Zhang, X.; et al. Safety and efficacy of mutant neoantigen-specific T-cell treatment combined anti-PD-1 therapy in stage IV solid tumors. Immunotherapy 2022, 14, 553–565.
- Cai, Z.; Su, X.; Qiu, L.; Li, Z.; Li, X.; Dong, X.; Wei, F.; Zhou, Y.; Luo, L.; Chen, G.; et al. Personalized neoantigen vaccine prevents postoperative recurrence in hepatocellular carcinoma patients with vascular invasion. Mol. Cancer 2021, 20, 164.
- Di Lorenzo, G.; Ferro, M.; Buonerba, C. Sipuleucel-T (Provenge®) for castration-resistant prostate cancer. BJU Int. 2012, 110, E99–E104.
- Lasek, W.; Zapała, Ł. Therapeutic metastatic prostate cancer vaccines: Lessons learnt from urologic oncology. Cent. Eur. J. Urol. 2021, 74, 300–307.
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422.
- Coupez, D.; Hulo, P.; Touchefeu, Y.; Bossard, C.; Bennouna, J. Pembrolizumab for the treatment of colorectal cancer. Expert Opin. Biol. Ther. 2020, 20, 219–226.
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410.
- Kudling, T.V.; Clubb, J.H.A.; Quixabeira, D.C.A.; Santos, J.M.; Havunen, R.; Kononov, A.; Heiniö, C.; Cervera-Carrascon, V.; Pakola, S.; Basnet, S.; et al. Local delivery of interleukin 7 with an oncolytic adenovirus activates tumor-infiltrating lymphocytes and causes tumor regression. Oncoimmunology 2022, 11, 2096572.
- Yao, Y.; Chen, H.; Tan, N. Cancer-cell-biomimetic nanoparticles systemically eliminate hypoxia tumors by synergistic chemotherapy and checkpoint blockade immunotherapy. Acta Pharm. Sin. B 2022, 12, 2103–2119.
- Li, C.; Liu, Y.; Li, D.; Wang, Q.; Zhou, S.; Zhang, H.; Wang, Y.; He, Z.; Liu, H.; Sun, J. Promising alternatives of CD47 monoclonal antibody: An injectable degradable hydrogel loaded with PQ912 for postoperative immunotherapy effectively blocks CD47-SIRPα signal. Theranostics 2022, 12, 4581–4598.
- Xie, W.; Chen, B.; Wen, H.; Xiao, P.; Wang, L.; Liu, W.; Wang, D.; Tang, B.Z. Biomimetic Nanoplatform Loading Type I Aggregation-Induced Emission Photosensitizer and Glutamine Blockade to Regulate Nutrient Partitioning for Enhancing Antitumor Immunotherapy. ACS Nano 2022, 16, 10742–10753.
- Xue, J.; Zhu, Y.; Bai, S.; He, C.; Du, G.; Zhang, Y.; Zhong, Y.; Chen, W.; Wang, H.; Sun, X. Nanoparticles with rough surface improve the therapeutic effect of photothermal immunotherapy against melanoma. Acta Pharm. Sin. B 2022, 12, 2934–2949.
- Li, T.; Liu, Z.; Fu, X.; Chen, Y.; Zhu, S.; Zhang, J. Co-delivery of Interleukin-12 and Doxorubicin Loaded Nano-delivery System for Enhanced Immunotherapy with Polarization toward M1-type Macrophages. Eur. J. Pharm. Biopharm. 2022, 177, 175–183.
- Xu, S.; Cui, F.; Huang, D.; Zhang, D.; Zhu, A.; Sun, X.; Cao, Y.; Ding, S.; Wang, Y.; Gao, E.; et al. PD-L1 monoclonal antibody-conjugated nanoparticles enhance drug delivery level and chemotherapy efficacy in gastric cancer cells. Int. J. Nanomed. 2018, 14, 17–32.
- Chen, Z.G. Small-molecule delivery by nanoparticles for anticancer therapy. Trends Mol. Med. 2010, 16, 594–602.
- George, R.; Hehlgans, S.; Fleischmann, M.; Rödel, C.; Fokas, E.; Rödel, F. Advances in nanotechnology-based platforms for survivin-targeted drug discovery. Expert Opin. Drug Discov. 2022, 17, 733–754.
- Li, L.; Zhang, Y.; Zhou, Y.; Hu, H.; Hu, Y.; Georgiades, C.; Mao, H.Q.; Selaru, F.M. Quaternary Nanoparticles Enable Sustained Release of Bortezomib for Hepatocellular Carcinoma. Hepatology 2022.
- Reda, M.; Ngamcherdtrakul, W.; Nelson, M.A.; Siriwon, N.; Wang, R.; Zaidan, H.Y.; Bejan, D.S.; Reda, S.; Hoang, N.H.; Crumrine, N.A.; et al. Development of a nanoparticle-based immunotherapy targeting PD-L1 and PLK1 for lung cancer treatment. Nat. Commun. 2022, 13, 4261.
- Rioja-Blanco, E.; Arroyo-Solera, I.; Álamo, P.; Casanova, I.; Gallardo, A.; Unzueta, U.; Serna, N.; Sánchez-García, L.; Quer, M.; Villaverde, A.; et al. Self-assembling protein nanocarrier for selective delivery of cytotoxic polypeptides to CXCR4(+) head and neck squamous cell carcinoma tumors. Acta Pharm. Sin. B 2022, 12, 2578–2591.
- Foglizzo, V.; Marchiò, S. Nanoparticles as Physically- and Biochemically-Tuned Drug Formulations for Cancers Therapy. Cancers 2022, 14, 2473.
- Wiklander, O.P.B.; Brennan, M.; Lötvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521.
- Yong, T.; Li, X.; Wei, Z.; Gan, L.; Yang, X. Extracellular vesicles-based drug delivery systems for cancer immunotherapy. J. Control. Release 2020, 328, 562–574.
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503.
- Yang, P.; Peng, Y.; Feng, Y.; Xu, Z.; Feng, P.; Cao, J.; Chen, Y.; Chen, X.; Cao, X.; Yang, Y.; et al. Immune Cell-Derived Extracellular Vesicles—New Strategies in Cancer Immunotherapy. Front. Immunol. 2021, 12, 771551.
- Hu, S.; Ma, J.; Su, C.; Chen, Y.; Shu, Y.; Qi, Z.; Zhang, B.; Shi, G.; Zhang, Y.; Zhang, Y.; et al. Engineered exosome-like nanovesicles suppress tumor growth by reprogramming tumor microenvironment and promoting tumor ferroptosis. Acta Biomater. 2021, 135, 567–581.
- Ma, Y.; Dong, S.; Li, X.; Kim, B.Y.S.; Yang, Z.; Jiang, W. Extracellular Vesicles: An Emerging Nanoplatform for Cancer Therapy. Front. Oncol. 2020, 10, 606906.
- Luo, F.Q.; Xu, W.; Zhang, J.Y.; Liu, R.; Huang, Y.C.; Xiao, C.; Du, J.Z. An Injectable Nanocomposite Hydrogel Improves Tumor Penetration and Cancer Treatment Efficacy. Acta Biomater. 2022, 147, 235–244.
- Kim, S.; Lee, J.; Im, S.; Kim, W.J. Injectable immunogel based on polymerized phenylboronic acid and mannan for cancer immunotherapy. J. Control. Release 2022, 345, 138–146.
- Nkanga, C.I.; Steinmetz, N.F. Injectable Hydrogel Containing Cowpea Mosaic Virus Nanoparticles Prevents Colon Cancer Growth. ACS Biomater. Sci. Eng. 2022, 8, 2518–2525.
- Santos, F.; Valderas-Gutiérrez, J.; Pérez Del Río, E.; Castellote-Borrell, M.; Rodriguez, X.R.; Veciana, J.; Ratera, I.; Guasch, J. Enhanced human T cell expansion with inverse opal hydrogels. Biomater. Sci. 2022, 10, 3730–3738.
- Wei, W.; Li, H.; Yin, C.; Tang, F. Research progress in the application of in situ hydrogel system in tumor treatment. Drug Deliv. 2020, 27, 460–468.
- Muraoka, D.; Harada, N.; Shiku, H.; Akiyoshi, K. Self-assembled polysaccharide nanogel delivery system for overcoming tumor immune resistance. J. Control. Release 2022, 347, 175–182.
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947.
- Kageyama, S.; Wada, H.; Muro, K.; Niwa, Y.; Ueda, S.; Miyata, H.; Takiguchi, S.; Sugino, S.H.; Miyahara, Y.; Ikeda, H.; et al. Dose-dependent effects of NY-ESO-1 protein vaccine complexed with cholesteryl pullulan (CHP-NY-ESO-1) on immune responses and survival benefits of esophageal cancer patients. J. Transl. Med. 2013, 11, 246.
- Schjetne, K.W.; Gundersen, H.T.; Iversen, J.G.; Thompson, K.M.; Bogen, B. Antibody-mediated delivery of antigen to chemokine receptors on antigen-presenting cells results in enhanced CD4+ T cell responses. Eur. J. Immunol. 2003, 33, 3101–3108.
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308.
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br. J. Cancer 2021, 124, 1759–1776.
- Carey, N.; Leahy, J.; Trela-Larsen, L.; McCullagh, L.; Barry, M. Tisagenlecleucel for relapsed/refractory acute lymphoblastic leukemia in the Irish healthcare setting: Cost-effectiveness and value of information analysis. Int. J. Technol. Assess. Health Care 2022, 38, e56.
- Talleur, A.C.; Myers, R.; Annesley, C.; Shalabi, H. Chimeric Antigen Receptor T-cell Therapy: Current Status and Clinical Outcomes in Pediatric Hematologic Malignancies. Hematol. Oncol. Clin. N. Am. 2022, 36, 701–727.
- Lyman, G.H.; Nguyen, A.; Snyder, S.; Gitlin, M.; Chung, K.C. Economic Evaluation of Chimeric Antigen Receptor T-Cell Therapy by Site of Care Among Patients with Relapsed or Refractory Large B-Cell Lymphoma. JAMA Netw. Open 2020, 3, e202072.
- Michels, A.; Ho, N.; Buchholz, C.J. Precision Medicine: In Vivo CAR Therapy as a Showcase for Receptor-Targeted Vector Platforms. Mol. Ther. 2022, 30, 2401–2415.
- Wang, Y.; Yang, P.; Zhao, X.; Gao, D.; Sun, N.; Tian, Z.; Ma, T.; Yang, Z. Multifunctional Cargo-Free Nanomedicine for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 2963.
This entry is offline, you can click here to edit this entry!