The dysfunction of the mismatch repair system, an important mechanism for the detection and correction of DNA replication mistakes, may often lead to instability in the length of specific genetic sequences, known as microsatellites, and to the accumulation of mutations. Microsatellite instability is a well-known risk factor for the development of colorectal cancers and other types of tumors but is also considered a positive predictor of the immunotherapy response. Malignancies harboring such a specific genomic instability are very immunogenic because of the great number of aberrant antigens they produce. Therapies based on the blockade of specific immune checkpoints have shown to induce an effective immune response against microsatellite-unstable cancer. Many studies proved that microsatellite instability has a decisive role in the carcinogenesis and the malignant progression of head and neck cancer and, in the near future, it may become a useful tool in tailoring immunotherapy also in this field of precision oncology.
- microsatellite instability
- MSI
- MMR proteins
- head and neck cancer
- progression
- local recurrence
- multiple primary tumors
- immunotherapy
1.Introduction
Head and neck cancer (HNC) accounts for 4–6% of all human solid malignancies and about 50% of patients ultimately die of the disease, despite recent improvements in HNC diagnosis and treatment [1]. In the last decade, a better knowledge of the complex interactions between cancer cells and the host immune system has led to novel immunotherapy strategies that, also in the setting of recurrent/metastatic HNC, have shown encouraging results in comparison to conventional treatment [2]. The development of immune checkpoint inhibitors, targeting PD-1 (programmed cell death-1), such as nivolumab and pembrolizumab, or PD-L1 (programmed death-ligand 1), such as atezolizumab and durvalumab, has dramatically changed the therapeutic scenario for several types of cancer, including melanoma, lung, breast, kidney and bladder. In HNC patients, these new drugs have shown to yield objective response rates ranging from 17 to 36% and, because of their direct and indirect costs and toxicities, the identification of the subgroup of patients that is most likely to benefit is of the uttermost importance [2][3]
Among the many possible biomarkers predictive of an immune response in HNC patients, recent reports have suggested a potential role of microsatellite instability (MSI) in the setting of several human solid malignancies [4]. We discuss the role of MMR alterations and the resulting MSI in HNC pathogenesis. Furthermore, by summarizing the clinical available data on how they influence the progression of precancerous lesions and the risk of recurrence or of developing a second primary tumor, we want to define the current role of MSI in the management of HNC patients.
2.MMR System and Development of MSI: An Overview
Microsatellites, also referred to as simple sequence repeats (SSRs), are short tandem DNA sequences (usually 1–5 nucleotide long) that are repeated from 5 to 100 times, and that are thought to account for approximately 3% of the full human genetic code [5]. There are many existent microsatellite polymorphisms that differ from person to person, and they are all characterized by a high degree of heterozygosity [6]. Usually, their lengths are very well conserved in the human genome but they are also particularly susceptible to inaccurate replication: the high rate of mutation in the number of repeats is mainly due to slippage phenomena of the DNA polymerase during the replication process [5]. If such an error occurs in healthy cells, it is promptly fixed by the mismatch repair system (MMR), a molecular machinery specialized in the detection and correction of replication mistakes, such as mispairing bases or small insertion/deletion loops (IDLs). In eukaryotic cells, the MMR system is composed of two different heterodimers: hMutS and hMutL [7]. The first one is the combination of the somatic protein hMSH2 (gene locus on 2p21) with either hMSH6 (2p16) or hMSH3 (5q14.1), resulting in hMutSα and hMutSβ heterodimers, respectively. On the other hand, hMutL is composed of hMLH1 (3p21) joined to hPMS2 (7q22.2), hPMS1 (2q31.1) or hMLH3 (14q24.3), resulting in hMutLα, hMutLβ and hMutLγ heterodimers, respectively [8]. The MSH2-MSH6 complex recognizes single base pair mismatches and 1–2 base IDLs (Figure 1.a), while MSH2-MSH3 primarily recognizes larger IDLs. To remove the mispaired DNA sequence, MutSα (or MutSβ) recruits MutLα (Figure 1.b), forming a tetrameric complex that proceeds to the excision step [9]: after PCNA (proliferating cell nuclear antigen, a component of the replication process) activates MutLα to incise the daughter strand far from the mismatched sequence (Figure 1.c), its progressive excision is performed by the MutSα-activated exonuclease EXO1 (Figure 1.d). Eventually, the DNA polymerases δ/ε carry out the DNA resynthesis [10](Figure 1.e).
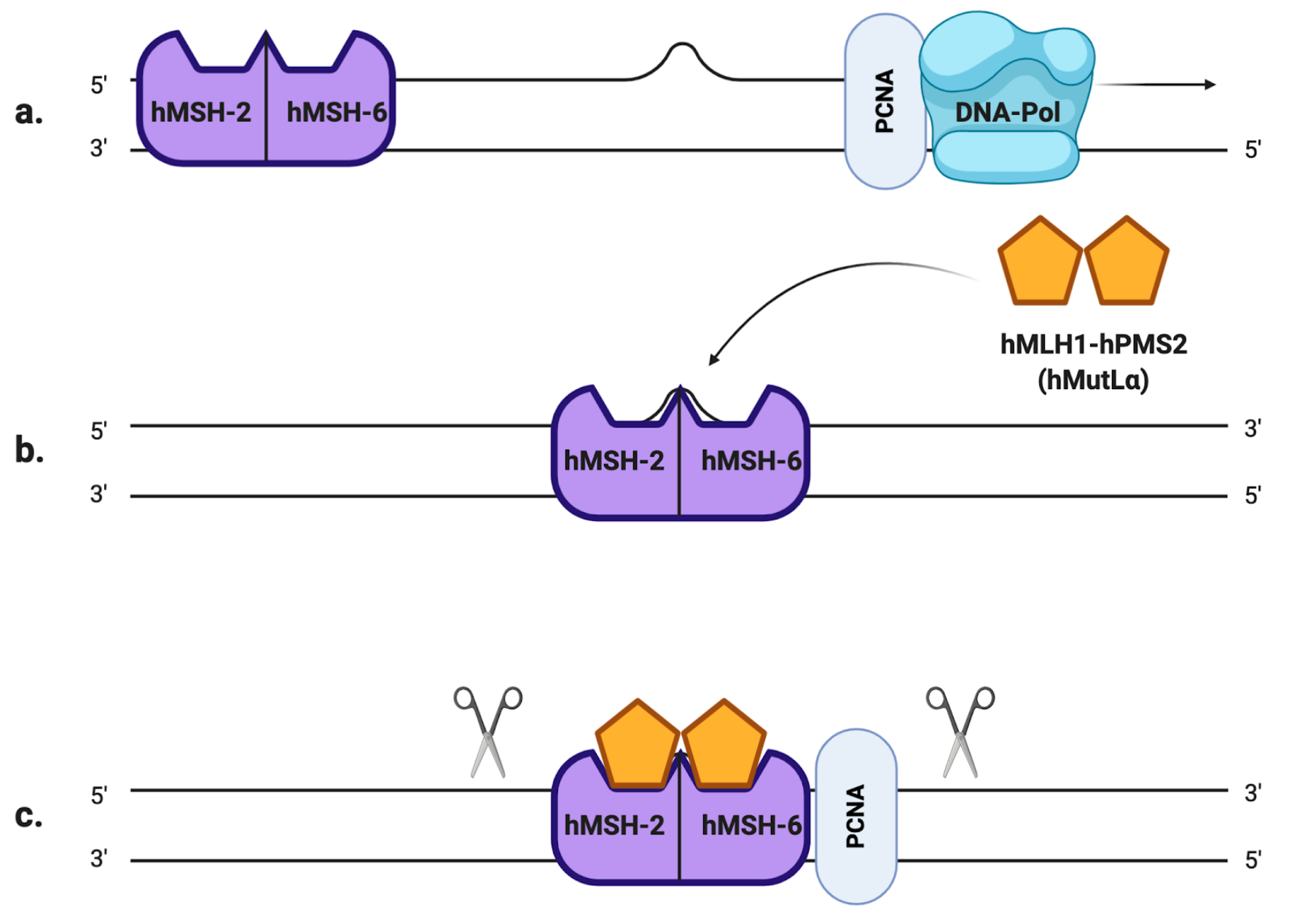
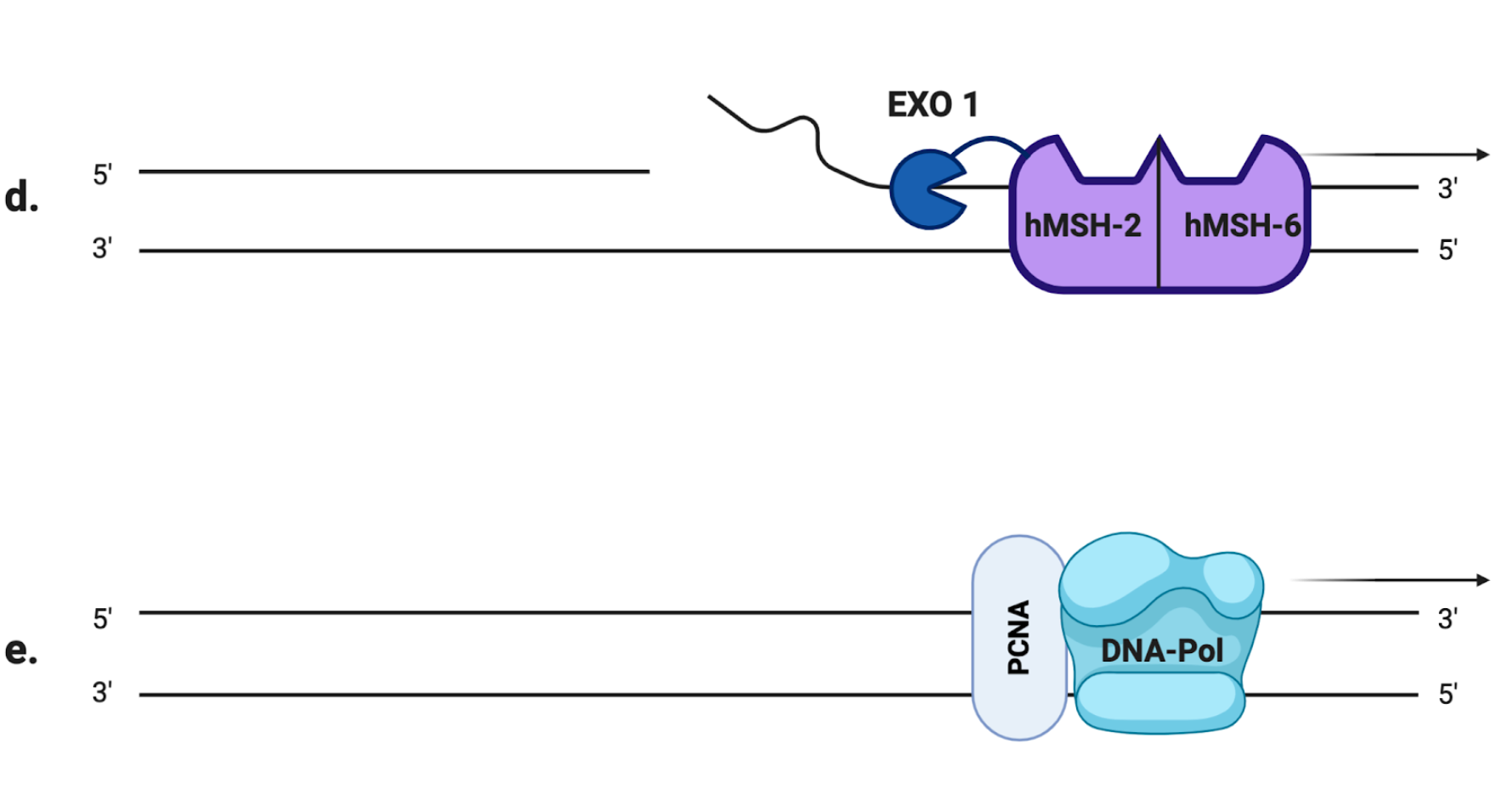
Figure 1. The mismatch repair (MMR) system (a) A mismatched base was wrongly incorporated in the newly synthesized DNA strand by DNA polymerase. (b) hMutSα detects the replication error and recruits hMutLα to form a heterodimer. (c) The newly synthesized strand is excised proximally and distally to the mismatched region by hMutLα, whose endonuclease activity is activated by PCNA. (d) hMutSα activates EXO1 that removes the excised region. (e) DNA polymerase carries out the DNA resynthesis.
The importance of the MMR in the replication process is proven by the fact that the error rate of DNA polymerases is estimated to be between 10−4 and 10−5 per nucleotide, whereas the activity of MMR can reduce the replication error-rate to 10−9–10−10 per nucleotide [11].
An altered function of the MMR mechanism can lead to the development of a so-called “mutator phenotype”. It is characterized by the accumulation of frameshift and missense mutations in many loci, including coding and non-coding microsatellite sequences, that become highly unstable in their lengths [12] (Figure 2). Interestingly, it seems that the altered function or depletion of MutSβ is related to a mutator phenotype less than the depletion of MutSα: this suggests that large IDLs arise less frequently or that they can be more easily repaired [11]. The resultant MSI implies a distinctive carcinogenic pathway associated with altered expression of specific tumor suppressor genes and the activation of oncogenes that can eventually lead to malignant transformation [13]. The same frameshift mutations that lead to the onset of MSI, if occurring in coding sequences, can cause the creation of specific tumor “neoantigens”, aberrant proteins that can be recognized by the immune system as “non-self” [11].
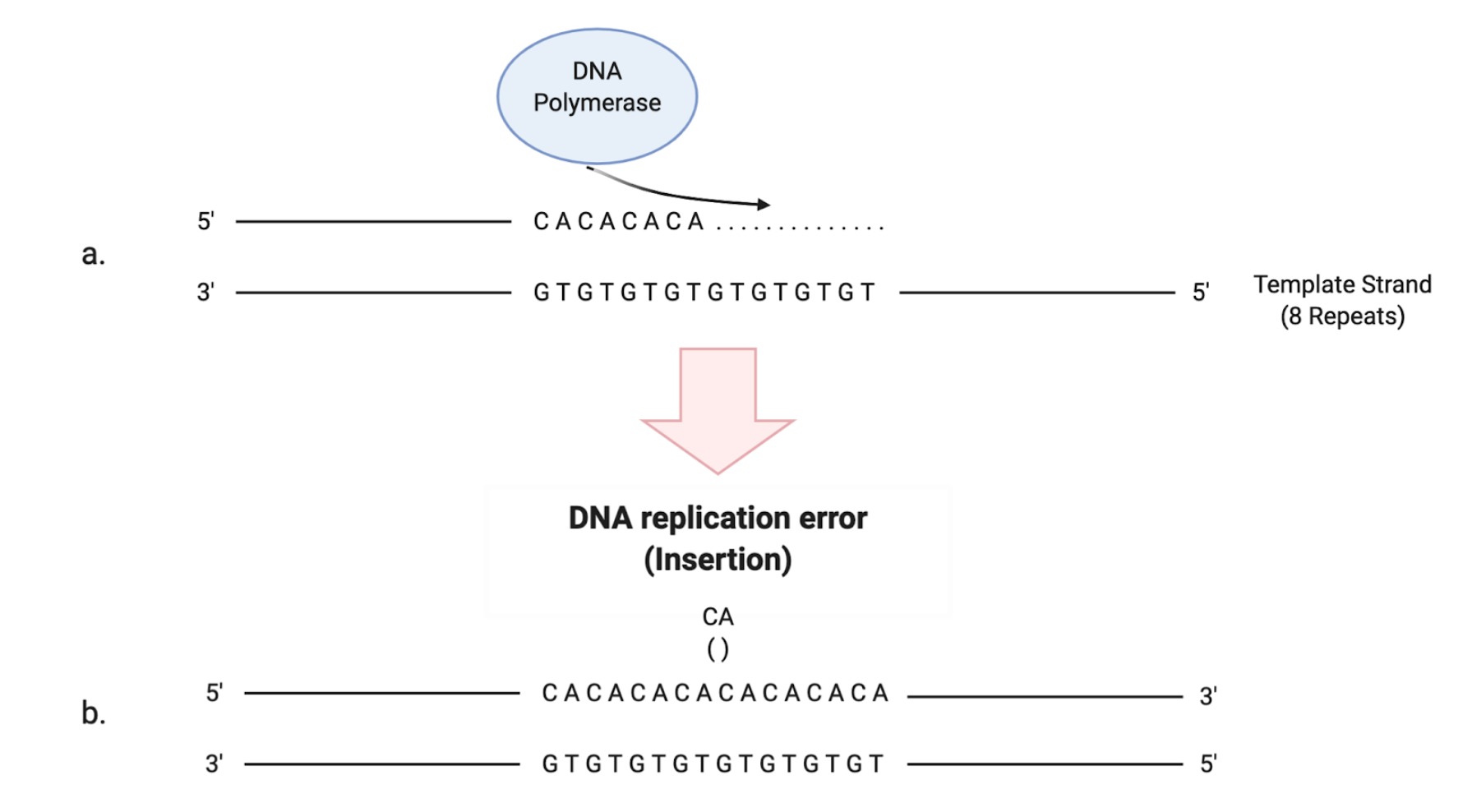
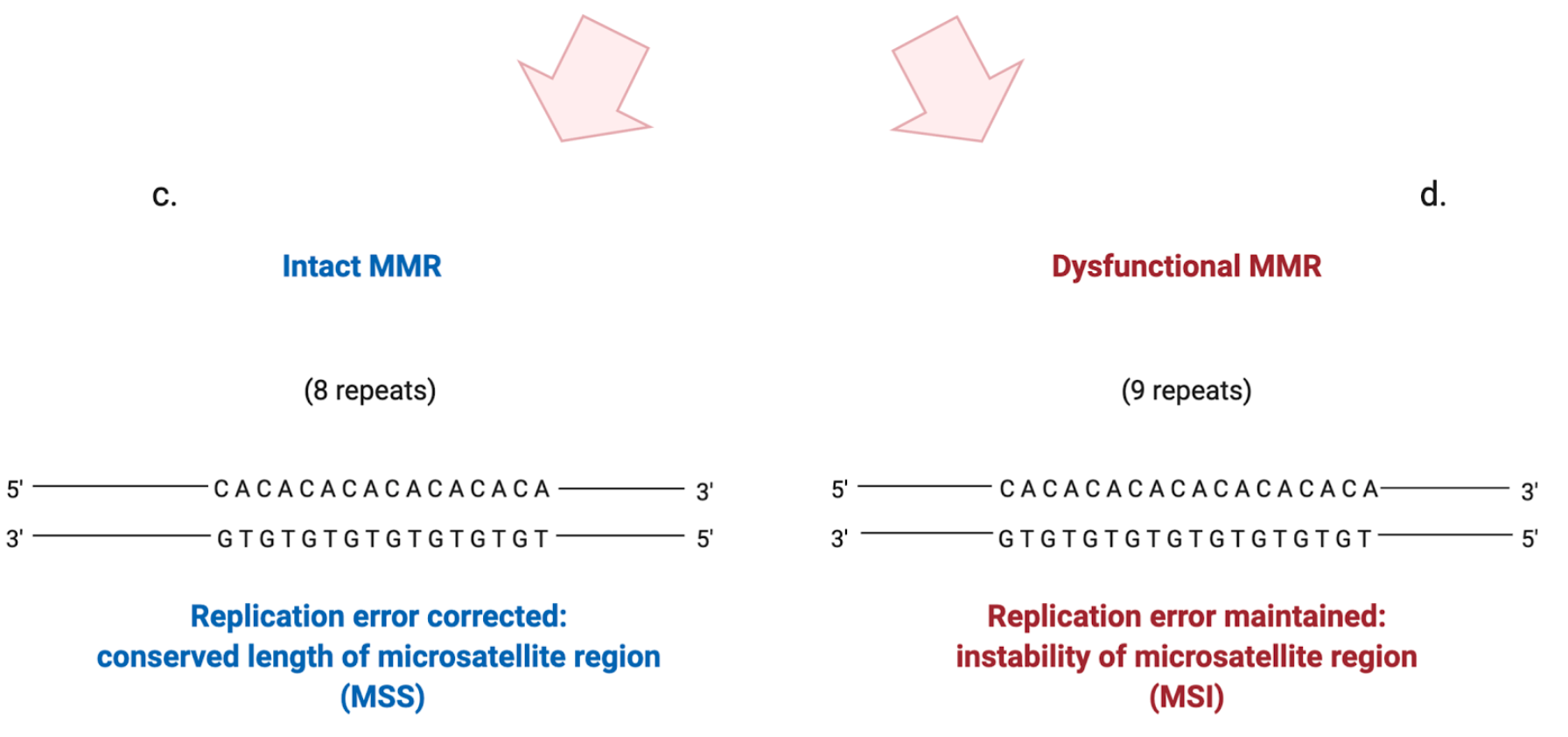
Figure 2. Schematic illustration of how the stability of microsatellite regions can be affected by the presence of a dysfunctional MMR system. (a) DNA polymerase performs the replication of a microsatellite sequence composed of 8 repeats of cytosine/adenine (CA). (b) Due to the slippage of DNA polymerase during replication, a CA repeat is wrongly incorporated. (c) If the MMR system is intact, the replication error is corrected and the right number of repeats is maintained. (d) If the MMR system is dysfunctional, the additional CA repeat will not be eliminated, leading to instability in the length of the microsatellite region.
MSI is historically considered to be the hallmark of one type of colorectal cancer (CRC) [14][15][16]. MSI was in fact discovered almost thirty years ago, when several research groups looking for molecular alterations or altered metabolic pathways that could be correlated to the development of CRC found that 10–15% of the sporadic cases were MSI+ [17][18]. Interestingly, they also observed that the presence of MSI could reach rates > 90% when testing patients affected by the Lynch syndrome (or HNPCC, hereditary non-polyposis colorectal carcinoma), the most common form of inherited cancer predisposition, and how this specific kind of genomic instability was related to the mutation or altered expression of DNA MMR-pathway proteins [6][19]. MSI+ sporadic CRC is usually related to the methylation of the promoter region of MLH-1, is predominantly located in the proximal colon and is poorly differentiated [6][18]. Often the susceptibility to chemotherapy, in particular to 5-FU, is altered: a few trials in the past concluded that fluorouracil-based adjuvant regimes did not show significant efficacy in the treatment of patients affected by highly unstable CRCs [19][20][21]. On the other hand, MSI+ CRC seems to have a higher radiation sensitivity, which is related to general genome instability with a direct implication of functional MMR proteins in DNA damage response induced by radiation [22]. Many studies also highlighted how MSI+ CRC is associated with increased survival and in general better prognosis in comparison to their genetic stable counterparts: they are in fact characterized by a lower incidence of distant metastasis and dense infiltrates of lymphocytes among the neoplastic tissue, a fact that deposes for an intense immune response against the neoplasm [6].
In 1998, the National Cancer Institute (NCI) proposed to test tumor and normal tissues using polymerase chain reaction (PCR) for five microsatellite markers as a standard procedure to detect MSI in CRC: two for mononucleotide repeats (BAT26 and BAT25) and three for dinucleotide repeats (D2S123, D5S346 and D17S250) [14]. The results of this analysis allow one to assess the grade of tumor MSI: MSI-high (MSI-H) for those with ≥ 30–40% marker instability (≥ 2 out of 5 markers); MSI-low (MSI-L) if marker instability is < 30–40% (1 out of 5 markers); microsatellite stable (MSS) if no instability is detected [23][24]. PCR-based testing is considered the gold standard in the detection of MSI, but it is an expensive procedure and it always needs the control testing of non-malignant tissues [25]. The alternative method is immunohistochemical staining (IHC) for altered MMR proteins, a technique that is cheaper and more easily available than PCR [25]. IHC has, however, an important limitation: it can detect only quantitative alterations in protein expression, not qualitative ones [14][25]. For example, a missense mutation in the MLH1 gene sequence would not alter the entity of its expression, yet it would produce a protein incapable of combining with PMS2 to form a functional heterodimer. To avoid the possible misinterpretation of MSI status and increase the sensitivity of IHC is recommended to perform the immunostaining of all the four principal MMR proteins: PMS2 and MSH6, in fact, are degraded when incapable of forming a heterodimer with functionally altered MLH1 or MLH2. The depletion of the former would therefore prove the qualitative alteration of the latter [26].
- MSI in Head and Neck Carcinogenesis
5.1. Impact on the Progression of Precancerous Lesions
In the last decades, many studies have been conducted to investigate the correlation between the development of MSI and HNC tumorigenesis [27].
Sengupta et al. [28] analyzed a group of patients affected by head and neck invasive or precancerous lesions: 50% of invasive squamous cell carcinoma (SCC) and 63% of dysplastic lesions showed promoter hypermethylation, a frequent epigenetic mechanism that inactivates gene transcription [29], of either or both hMLH1 and hMSH2. At least for the preneoplastic subset, the number of samples harboring MMR epigenetic inactivation was shown to be related to the grade of MSI detected, given that the hypermethylation of the MMR promoter grew proportionally with the degree of MSI. They also showed that tobacco use increased susceptibility to hMLH1 and hMSH2 hypermethylation not only in the affected lesion but also in the adjacent unaffected epithelium [28].
In 2011 Caldeira PC et al. [30][31] investigated this topic further and carried out two different studies demonstrating how hMLH1 expression decreased from low-grade oral leukoplakias to lesions showing severe dysplasia. Further studies highlighted the significant association of hMSH3 mutations with the development of leukoplakia and its progression to OSCC and that a decreasing trend in MMR expression can be detected when testing non-dysplastic samples and oral preneoplastic lesions with progressively increasing severity [32][33]. A lot of research was done on how the disruption of the MMR system is related to the development of OSCC: Ha PK et al. [34] described, in a cohort of 93 premalignant lesions of the upper aerodigestive tract and 18 invasive OSCC, a trend toward a higher MSI rate according to the histopathological severity of lesions: 6% in the hyperplastic lesions without atypia, 27% in the severe dysplasia/CIS and 33% in the invasive cancers.
The detection of MSI in OSCC samples seems to be significantly associated with the tumor stage, differentiation grade and the tendency to develop multiple oral malignancies [35][36][37].
As mentioned before, the decreased expression of MMR proteins, and hMLH1 in particular, can also be caused by promoter hypermethylation. In a matched case-control study, including 50 OSCC cases and 200 controls, promoter methylation of hMLH1 was detected (using methylation-specific PCR) in 38 (76%) of the cancers, but in none of the control samples. All the OSCC that harbored hMLH1 epigenetic alteration and tested negative for hMLH1 immunostaining were diagnosed at an early stage, thus reinforcing the concept that hMLH1 promoter hypermethylation seems to occur at the beginning of the OSCC carcinogenic pathway [38]. However, when considering the results of this particular study, it should be reminded that, although being a cheap and highly sensitive method for the DNA methylation quantification, methylation-specific PCR is burdened by a high rate of false-positive results [39].
Helal et al. [40] used immunohistochemistry to compare the pattern of expression of hMSH2 proteins between OSCC and oral dysplastic lesions (DL). Reduced expression of hMSH2 was detected in 26 of the 70 oral SCC (37.1%) and 2 of 21 oral DL (9.5%) with a statistically significant difference (p = 0.03), suggesting that the expression of the MMR system proteins diminishes as the histological severity of the lesion increases. These results were later confirmed by other authors, reinforcing the hypothesis of a direct correlation between MMR protein expression and dysplasia grade[41].
It seems that even inherited abnormalities of MMR protein genes can be associated with an increased risk or worse outcomes for HNC patients, particularly among smokers [42]. A study that genotyped 242 patients with tobacco-related OSCC and 205 healthy controls by polymerase chain reaction–restriction fragment length polymorphism (PCR–RFLP) technique, explored the risk related to the presence of the hMLH1 -93 A > G (rs 1800734) single nucleotide polymorphism (SNP). It resulted that this specific polymorphism is associated with a higher risk of tobacco-related OSCC and could represent a useful screening marker [43]. The sample size of this study is quite low: even though a significant association of this specific SNP has been detected also for colorectal, lung and endometrial cancers [44][45][46], future larger studies (e.g., GWAS stratified by MSI status) are needed to corroborate these results.
The malfunction of MMR and the reduced expression of its proteins not only correlate with the progression from preneoplastic lesions to OSCC but also with the stage and the behavior of oral invasive malignancies. High hMSH2 (Figure 3 B.a) and hMLH1 expression (Figure 3 A.a) have been associated with a reduced depth of invasion, and the absence of perineural invasion [47], while MMR deficient tumors (Figure 3 A.b and B.b) have shown higher rates of bone invasion and high pT stage, and the presence of metachronous neoplasms [48].
Among the articles analyzed, only the paper by Pimenta et al. considered oral lichen planus (OLP): the actual risk for malignant transformation of OLP is controversial, as just some studies support its premalignant nature [49][50]. The actual overall frequency of OLP malignant transformation is estimated to be between 0.3 and 12.5% [49][50]. They examined the expression of hMSH2 protein by immunohistochemistry in 26 cases of OLP and in 10 samples of non-malignant mucosa as a control group. The percentage of cells expressing hMSH2 in reticular and atrophic/erosive subtypes of OLP was lower (46.54% and 48.79%, respectively) compared to normal mucosa (61.29%). It is possible that the reduced expression of the hMSH2 protein makes the epithelium of OLP more susceptible to DNA mutation, making it prone to OSCC development [51]
Some of the papers dealt with the process of carcinogenesis in the lips and its relation to MMR dysfunctions: Souza et al. [52] tested (in normal, dysplastic and malignant lip epithelium) the tissue expression of hMSH2 and other DNA repair proteins (p53, a most important tumor suppressor gene, APE1, a multifunctional enzyme of the base excision repair pathway, and ERCC1, a component of the nucleotide excision repair mechanism), finding a significant reduction in epithelial cell expression of these proteins. Other authors observed a decrease in the immunohistochemical determination of hMLH1 and hMSH2 in line with the increasing histologic grade. The largest number of MSI+ cells was detected in biopsies of actinic cheilitis without or with mild dysplasia, intermediate values were obtained for lesions with moderate/severe dysplasia and the lowest number of positive cells was presented by lower lip SCC [53][33][54].
Moving to other anatomical districts, Yalniz et al. [55] found that in their group of 99 patients (65 affected by laryngeal cancer) 26 were positive for MSI, with 17 patients displaying instability at more than one locus (MSI-H). In 2006 Demokan et al. [56] observed even higher rates. Among a cohort of 116 patients with many types of HNC (85 larynx, 13 salivary glands, 10 oral cavity, 6 nasal/paranasal sinus and 1 glomus), MSI was detected in the 41% of the samples and in the 59% of cases the promoter region of hMLH1 or hMSH2 resulted hypermethylated.
The development of MSI seems to be an indicator of a higher risk of malignant progression also in the larynx. Dysplastic laryngeal lesions from patients already affected by laryngeal cancer have significantly higher rates of MSI in comparison to preneoplastic lesions from otherwise healthy patients [57].
The presence of MSI and its relation with malignant evolution was also studied for the most frequent benign salivary gland tumor, pleomorphic adenoma (PA), with a malignant transformation rate around 3–4%. Despite the presence of a hyalinized stroma and focal calcifications are proven to be predictive for the risk of cancer development, a lot of different histologic features are only considered suggestive of such a risk, including cytologic atypia, increased mitoses and invasion of the capsule [58][59]. Researching for a useful immunohistochemical marker, Tobón-Arroyave et al. [60] conducted a study to examine the relationship between all those carcinogenesis-related histopathological features and the immunoexpression of hMLH1 and hMSH2 proteins in a group of 35 benign PA lesions, divided into low- and a high-risk subtype. Their findings were coherent with other papers that dealt with preneoplastic lesions of different head and neck districts: the mean expression values of both hMLH1 and hMSH2 were significantly lower in the high-risk subtype.
While the majority of authors agree in recognizing the significant prevalence of MSI and MMR dysfunctions in HNC and their role in neoplastic progression, few groups have obtained opposite results: De Schutter et al. in 2009 have, for example, concluded that MMR deficiency contributes little to the carcinogenic process of HNC, reporting that just one out of the eighty patients enrolled in their study was found positive for MSI [61].
Similar findings were obtained in two articles investigating MSI prevalence and hMSH2/hMLH1 altered expression in salivary gland tumors, either benign or malignant: a low-frequency microsatellite instability and no significant hMSH2/hMLH1 expression difference between benign and malignant neoplasms were detected [62][63].
A Canadian study was instead able to find a significant correlation between HNSCC and the presence of MSI, but not with MMR proteins dysfunction: investigating MSI status and hMLH1 and hMSH2 expression among 24 young HNSCC patients (≤ 44 years old; 46% smokers) and comparing it with an older cohort (33 HNSCC cases, ≥ 45 years, 88% with a history of tobacco abuse), they found that 100% of tumors in the younger cohort were MSI+ at least at one site, while 61% MSI+ tumors were found among the older patients [64]. The authors showed MSI to be more frequent in the non-smoker group and that it could be mainly detected in early-stage HNC, suggesting that genomic instability does occur in the early phases of tumor initiation/progression; however, they could not find any correlation between the inactivation of hMLH1 and hMSH2 genes and the development of MSI [64].
Of course, some of the included studies were limited by the small size of their cohorts of patients; furthermore, we have to consider that the anatomopathological methods to assess microsatellite instability and MMR protein expression are far from being standardized. The aforementioned methodological pitfalls of microsatellite markers-PCR and MMR IHC staining must not be underestimated; nonetheless, the consistency and the statistical significance of all these results cannot be ignored.
5.2. Impact on Local Recurrence
An interesting application of MSI-determination is the one performed on histopathologically tumor-free surgical margins [58][65]. Based on the assumption that the detection of instability in the microsatellite sequences length underlies a genotype able to trigger the malignant transformation, a few authors focused on the investigation of MSI markers on the negative margins they obtained after performing “radical” surgical resection of head and neck cancers, in order to see if it could represent a significant predictor of the risk of local recurrence.
As early as 2000, Sardi et al. used PCR to assess the presence of frame-shift alterations or loss of heterozygosity (LOH) of 10 different microsatellite markers in the surgical margins of a cohort of 41 patients affected by HNC. Among the 25 samples that resulted tumor-negative after histopathological analysis, 11 showed the same microsatellite alterations as in the primary tumors: seven of those patients experienced a local recurrence, while just one of the 14 tumors with MSI-negative margins recurred. Performing regression analysis the authors showed how molecularly positive margins were in fact an independent prognostic factor for local recurrence (p = 0.04) [58].
A study with a similar design tested five different tetranucleotide microsatellite markers on tumor samples and corresponding surgical margins from 54 patients who had undergone surgery for HNC and whose margins resulted in a microscopically negative status. Twenty-six (48%) tumors were positive for MSI and seven (27%) out of them had the same instability pattern in the paired margins. Of those seven patients, five developed a local relapse, suggesting an independent association between detection of MSI in the surgical margins and local recurrence of the tumor [65].
More recently, two other studies have tested a large cohort of OSCC cases using microsatellite markers on histopathologically negative surgical margins. The same conclusions were reached: the detection of microsatellite instability in tumor-free margins was related in a statistically significant manner to a higher risk of local recurrence [66][67].
All these studies ultimately suggest that molecular assessment of surgical margins’ microsatellite status could be a useful tool in HNC patients’ management to identify the subgroup that needs a more intense follow-up.
5.3. MSI as a Risk Factor for Multiple Primary Cancers
It is well known how several risk factors, such as tobacco and alcohol consumption or human papillomavirus (HPV) infection, have been associated with the development of multiple mutations and carcinogenic genotype in wide areas of tissue that can lead to disease progression, cancer relapse and multiple primary malignancies, according to a “field cancerization” model [68][69]. In addition, genetic factors may contribute to the development of a mutator phenotype, and molecular markers of genetic instability, such as MSI markers, could be a useful tool to identify patients particularly susceptible to the development of multiple primary cancers (MPC) in these districts [70].
A recent review [71] investigated this topic, focusing on various inherited and acquired gene mutations (germ-line mutations, single-nucleotide polymorphism, chromosomal instability, microsatellite instability and DNA methylation) in head and neck malignancies and researching the literature to find if each of them could have a correlation with increased susceptibility to MPCs. For MSI the authors considered just one study, performed in 1998 by Piccinin et al., in which MSI was analyzed at 20 chromosomal loci in 67 HNSCC patients (45 with single cancer and 22 with multiple primary tumors). The 22 MPCs cases were also searched for hMLH1 gene mutations, to evaluate its possible involvement in MSI development. They found neither a significant difference in the frequency of MSI comparing MPCs and single cancer cases nor somatic or germline mutations of the hMLH1 gene in microsatellite instable neoplasms [72].
Other studies reached different conclusions: in 2005 an Italian team enrolled 32 patients suffering from a new tumor on the “field” of a previously surgically treated oral or oropharyngeal carcinoma. They tested a panel of eight different microsatellite markers that were specifically selected to investigate the status of oncogenes/tumor suppressor genes such as p16 and p53. They retrieved samples from both the primary tumor and the newly developed one, finding that only eight of them showed an identical clonal pattern of microsatellite abnormalities, confirming that the more recent tumor was an actual recurrence of the index one. On the other hand, 15 patients (46,9%) showed different patterns of MSI in the two samples: the different genetic arrangement suggests an independent origin of the tumor, yet it supports the idea of a “field cancerization” related to genomic instability that induces the development of new genetic mutations and, as a consequence, second primary tumors [69][73].
More recently, other authors obtained results that seem to confirm this hypothesis: the detection of MSI in the index tumor has been found to have a statistically significant correlation to the risk of developing a second primary tumor in a large cohort of HNC patients [74].
Finally, epigenetic alterations of MMR genes, whose dysfunctions are well known to be related to genomic instability, has shown a positive correlation with the risk of MPC: Czerninski et al. demonstrated how, in their cohort of OSCC, the 100% of patients affected by multiple oral malignancies tested positive for hypermethylation of hMLH1 or hMSH2 promoters [36].
- MSI and Immunotherapy of HNC
HNCs are frequently diagnosed when already at stage III/IV of the disease and the standard therapy consists of the association of surgery, radiation and/or chemotherapy. Nevertheless, the average failure rates remain considerable (60% locoregional and 30% distant recurrence rate) [75]. Platinum-based chemotherapy is the regimen most commonly used and the development of biological resistance to this type of drug is a well-known issue in this setting. A study performed on cisplatin-resistant vs. cisplatin-sensitive HNSCC cell lines reported that the former had a significantly decreased expression of MLH1 compared to the latter [76], confirming previous evidence suggesting that a dysfunctional MMR is associated with a worse response to this type of chemotherapy [77][78]. The specific underlying mechanism of the MMR-deficiency related chemoresistance is yet to be clarified: most likely, the dysfunctional MMR acts as a component in a complex association with other genetic alterations, such as defective p53-mediated DNA damage response pathway, cell-death controlling factors or other mutations that determine a diminished drug uptake in cancer cells [79].
Searching for new and effective HNC treatment options, an increasingly important role is being attributed to immunotherapy, that has recently revolutionized a large part of the oncology practice: the assumption on which it is based is that the human immune system has the innate ability to recognize cancer cells through specific “neoantigens” that are the results of the transcription of mutated DNA sequences generated during the process of malignant transformation [80]. This protective mechanism is usually evaded by tumors through the acquisition of immunoresistance and multiple checkpoints pathways, which normally help to maintain self-tolerance, and can be exploited by cancer cells to evade immune surveillance [81]. The programmed cell death protein 1 (PD-1) has emerged in the last years as one of the most important of these inhibitory pathways, due to its role in regulating the activity of T cells [82]. The first reports of cancer overexpressing this transmembrane glycoprotein and its ligand (PD-L1) date back to 2002 on a mice population: PD1-high tumor cells showed enhanced invasiveness and they were immune to cytotoxic T cells lysis, but both effects resulted reversible by the administration of anti-PD-L1 antibodies [83]. It has been observed how highly genetically unstable tumors, such as the ones that developed the aforementioned “mutator phenotype” (MSI and a high number of deletion/insertion frameshift mutations), are the most susceptible to this type of therapy [84].
MSI+ cancers are very immunogenic and are usually highly infiltrated with activated CD8+ cytotoxic T and T helper 1 lymphocytes, actively producing IFN-γ [85]. Interestingly interferons (IFNs), and IFN-γ in particular, happen to be the principal up-regulator of the expression of multiple immune checkpoint inhibitors, including the PD-1/PD-L1 pathway [86][85]. Summing up, the most immunogenic tumors seem to be the ones with the strongest inhibition of the innate immune response [86].
In recent years, many studies demonstrated that MMR deficiency and MSI could predict the tumor response to immunotherapies consisting of the administration of anti-PD-1 (pembrolizumab and nivolumab) and anti-PD-L1 (atezolizumab and durvalumab) antibodies. D.T. Le et al., in two subsequent studies, showed how immune checkpoint blockade with pembrolizumab induced an objective radiographic response in 40–53% patients with MMR-deficient cancers originated from various organs, in contrast to MMR-proficient malignancies, which did not benefit from the therapy [4][87]. It seems that, in particular, an high load of insertion/deletion-related frameshift mutations, typically associated with an MMR dysfunction, is highly predictive of a good anti-PD-1 immunotherapy response and that, for MSI+ tumors, the clinical benefit of immune checkpoint blockade directly correlates with the intensity of MSI (MSI-H, MSI-L or MSS) [88].
Although the efficacy of checkpoint blockade therapies and its correlation to genomic instability have been thoroughly studied predominantly for colorectal cancer, anti-PD-1 immunotherapies have also been approved as a second-line treatment for other types of advanced/metastatic malignancies [82].
The use of pembrolizumab and nivolumab, in particular, was approved in 2016 by the Food and Drug Administration (FDA) for the treatment of recurrent or metastatic (R/M) HNSCC associated with disease progression after the first-line treatment, which commonly consists in a combination of 5-FU, cisplatin/carboplatin and cetuximab (EXTREME regimen) [89][90]. More recently, pembrolizumab has been approved by the FDA as a first-line treatment itself in R/M HNSCC: in monotherapy (only in patients expressing high rates of PD-L1) or in combination with standard chemotherapeutic agents [3].
Many trials have been carried out to determine the effective efficacy of anti-PD-1 therapies in advanced HN cancers: the Phase Ib trial KEYNOTE-012, in which pembrolizumab was administered to 60 patients affected by R/M HNSCC, resulted in an overall response rate (ORR) of 18%, 25% in human papillomavirus (HPV)-positive and 14% in HPV-negative patients [91]. It seems that HPV viral gene products could increase the immunogenicity of the tumor, thus improving the efficacy of the immune checkpoint blockade [89]. All the patients enrolled in that trial had PD-L1-positive immunostaining in at least 1% of cancer, stromal or inflammatory cells that constituted the tumor microenvironment [89]. In order to better understand the possible role of PD-L1 expression as a predictive biomarker for immune checkpoint blockade therapy in HN cancers, the follow-up expansion cohort of the KEYNOTE-012 allowed enrollment regardless of the PD-L1 expression status [92]. Furthermore, the authors chose to compare two different scoring methods to evaluate the PD-L1 IHC assay: the tumor proportion score (TPS), which is the percentage of viable tumor cells showing partial or complete membrane staining at any intensity, and the combined positive score (CPS), the number of PD-L1 staining cells (tumor cells, lymphocytes and macrophages) divided by the total number of viable tumor cells, multiplied by 100 [93]. When sorted using TPS, no significant difference in terms of response to pembrolizumab was found between positive (≥ 1%) and negative (< 1%) PD-L1 expression tumors. Conversely, when the whole tumor microenvironment was considered by CPS, a statistically significant increase in the response rate to immunotherapy was observed: CPS ≥ 1% patients had an ORR of 22%, in contrast with the 4% ORR of the negative ones (CPS < 1%) [92].
A recent clinical review collected the results of twelve studies in which PD-1/PD-L1 inhibitors were administered to a total of 1088 R/M HNSCC patients: 93% of them were tested for PD-L1 expression rate (with different scoring methods) and 67% for the HPV status. The meta-analysis of the outcomes seems to confirm that higher rates of PD-L1 expression enhance the overall response rate (ORR) of these patients: 18.9% for expressers versus 8.8% for non-expressers. On the other hand, in contrast to previous suggestions, no differences were observed in survival or tumor response among the HPV positive patients [94].
It is important to highlight that, even if the choice of CPS as a scoring method to evaluate the PD-L1 expression seems to strengthen its predictive potential [92], the results of the most recent trials where pembrolizumab was administered to R/M HNSCC patients seem to suggest that anti-PD-L1 immunotherapy should not be restricted only to PD-L1 expressing tumors. The authors of Phase II study KEYNOTE-055 [95] in fact, although observing higher response rates in patients with higher PD-L1 expression, reported a clinically meaningful response also in those that tested negative to immunostaining (18% ORR in CPS ≥ 1% vs. 12% ORR in CPS < 1% patients). Furthermore, 6- and 12-month progression-free survival and overall survival rates were similar in the two groups.
Similar conclusions were reached in the Phase III study KEYNOTE-048 [3]: 882 participants affected by untreated locally incurable R/M HNSCC, stratified by performance status, p16 status and PD-L1 CPS, were randomized in three groups and received, respectively, pembrolizumab alone, pembrolizumab with chemotherapy (platinum and 5-fluorouracil) and cetuximab with chemotherapy. The results demonstrated that pembrolizumab monotherapy significantly improved overall survival (OS) in PD-L1 CPS ≥ 1% patients while having the non-inferior OS, a longer duration of response and a better safety profile when compared to cetuximab with chemotherapy. Moreover, pembrolizumab with chemotherapy significantly improved OS, prolonged duration of response and had a similar safety profile versus cetuximab with chemotherapy in the whole population [3].
In conclusion, while PD-L1 CPS ≥ 1% appears to be a useful biomarker of favorable clinical outcomes of immunotherapy in HN cancers, the lack of its expression does not seem to be a consistent negative predictor of response to the immune checkpoint blockade. A recent case report seems to confirm this hypothesis but also opens up to new interesting possibilities: a 62-year-old man presenting an advanced SCC (T4N2M0) of the right piriform sinus that underwent standard induction chemotherapy followed by platinum-based radiochemotherapy, pharyngo-laryngectomy and unilateral cervical dissection for a first local recurrence and stereotactic reirradiation for a second one. A third loco-regional recurrence was found to be inaccessible neither by surgery nor radiotherapy. The patient was eventually enrolled in a Phase II trial and underwent the administration of anti-PD-L1. After five infusions, a complete clinical and radiological response was observed, despite the immunohistochemical PD-L1 expression status being proven to be negative. Interestingly, PCR and IHC performed on tumor samples demonstrated the presence of MSI-H, MLH1 and PMS2 expression loss. Those findings suggest that the same “mutator phenotype” associated with objective response to immune checkpoint blockade in CRC could be found in HN cancers as well and that MSI and MMR impairment could possibly be used as a predictor of immunotherapy outcomes [96].
This entry is adapted from the peer-reviewed paper 10.3390/cancers12103006
References
- Laura Q.M. Chow; Head and Neck Cancer. New England Journal of Medicine 2020, 382, 60-72, 10.1056/nejmra1715715.
- John D. Cramer; Barbara Burtness; Robert L. Ferris; Immunotherapy for head and neck cancer: Recent advances and future directions. Oral Oncology 2019, 99, 104460, 10.1016/j.oraloncology.2019.104460.
- Barbara Burtness; Kevin J Harrington; Richard Greil; Denis Soulières; Makoto Tahara; Gilberto De Castro; Amanda Psyrri; Neus Basté; Prakash Neupane; Åse Bratland; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. The Lancet 2019, 394, 1915-1928, 10.1016/s0140-6736(19)32591-7.
- Dung T. Le; Jennifer N. Durham; Kellie N. Smith; Hao Wang; Bjarne R. Bartlett; Laveet K. Aulakh; Steve Lu; Holly Kemberling; Cara Wilt; Brandon S. Luber; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409-413, 10.1126/science.aan6733.
- Hans Ellegren; Microsatellite evolution: a battle between replication slippage and point mutation. Trends in Genetics 2002, 18, 70, 10.1016/s0168-9525(02)02631-8.
- Karl Heinimann; Toward a Molecular Classification of Colorectal Cancer: The Role of Microsatellite Instability Status. Frontiers in Oncology 2013, 3, 272, 10.3389/fonc.2013.00272.
- Flora S. Groothuizen; Titia K. Sixma; The conserved molecular machinery in DNA mismatch repair enzyme structures. DNA Repair 2016, 38, 14-23, 10.1016/j.dnarep.2015.11.012.
- Gleyson Kleber Do Amaral‐Silva; Manoela Domingues Martins; Hélder Antônio Rebelo Pontes; Eduardo Rodrigues Fregnani; Márcio Ajudarte Lopes; Felipe Paiva Fonseca; Pablo Agustin Vargas; Mismatch repair system proteins in oral benign and malignant lesions. Journal of Oral Pathology and Medicine 2016, 46, 241-245, 10.1111/jop.12484.
- Dipika Gupta; Christopher D. Heinen; The mismatch repair-dependent DNA damage response: Mechanisms and implications. DNA Repair 2019, 78, 60-69, 10.1016/j.dnarep.2019.03.009.
- Dorothy A. Erie; Keith R. Weninger; Single molecule studies of DNA mismatch repair. DNA Repair 2014, 20, 71-81, 10.1016/j.dnarep.2014.03.007.
- Dekang Liu; Guido Keijzers; Lene J. Rasmussen; DNA mismatch repair and its many roles in eukaryotic cells. Mutation Research/Reviews in Mutation Research 2017, 773, 174-187, 10.1016/j.mrrev.2017.07.001.
- Lawrence A. Loeb; A mutator phenotype in cancer.. Cancer Research 2001, 61, 3230-3239, .
- Jason H. Bielas; Keith R. Loeb; Brian P. Rubin; Lawrence D. True; Lawrence A. Loeb; Human cancers express a mutator phenotype. Proceedings of the National Academy of Sciences 2006, 103, 18238-18242, 10.1073/pnas.0607057103.
- Camille Evrard; Gaëlle Tachon; Violaine Randrian; Lucie Karayan-Tapon; David Tougeron; Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer. Cancers 2019, 11, 1567, 10.3390/cancers11101567.
- Sanjay Popat; R. Hubner; R.S. Houlston; Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. Journal of Clinical Oncology 2005, 23, 609-618, 10.1200/jco.2005.01.086.
- Fabio Gelsomino; Monica Barbolini; Andrea Spallanzani; Giuseppe Pugliese; Stefano Cascinu; The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treatment Reviews 2016, 51, 19-26, 10.1016/j.ctrv.2016.10.005.
- Bharati Bapat; Noralane M. Lindor; John Baron; Kim Siegmund; Lin Li; Yingye Zheng; Robert Haile; Steve Gallinger; Jeremy R. Jass; Joanne P. Young; et al. The Association of Tumor Microsatellite Instability Phenotype with Family History of Colorectal Cancer. Cancer Epidemiology Biomarkers & Prevention 2009, 18, 967-975, 10.1158/1055-9965.epi-08-0878.
- Frank A. Sinicrope; Lynch Syndrome–Associated Colorectal Cancer. New England Journal of Medicine 2018, 379, 764-773, 10.1056/nejmcp1714533.
- G. Kurzawski; J. Suchy; T Dębniak; J. Kładny; J. Lubinski; Importance of microsatellite instability (MSI) in colorectal cancer: MSI as a diagnostic tool. Annals of Oncology 2004, 15, iv283-iv284, 10.1093/annonc/mdh940.
- Daniel J. Sargent; Silvia Marsoni; Genevieve Monges; Stephen N. Thibodeau; Roberto Labianca; Stanley R. Hamilton; Amy J. French; Brian Kabat; Nathan R. Foster; Valter Torri; et al. Defective Mismatch Repair As a Predictive Marker for Lack of Efficacy of Fluorouracil-Based Adjuvant Therapy in Colon Cancer. Journal of Clinical Oncology 2010, 28, 3219-3226, 10.1200/jco.2009.27.1825.
- M. M. Bertagnolli; C. C. Compton; D. Niedzwiecki; R. S. Warren; S. Jewell; G. P. Bailey; R. J. Mayer; R. Goldberg; L. Saltz; M. Redston; et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, 5-fluorouracil and leucovorin in stage III colon cancer. Journal of Clinical Oncology 2006, 24, 10003-10003, 10.1200/jco.2006.24.18_suppl.10003.
- Joo-Shik Shin; Thein Ga Tut; Tao Yang; C. Soon Lee; Radiotherapy Response in Microsatellite Instability Related Rectal Cancer. The Korean Journal of Pathology 2013, 47, 1-8, 10.4132/koreanjpathol.2013.47.1.1.
- Marina Baretti; Dung T. Le; DNA mismatch repair in cancer. Pharmacology & Therapeutics 2018, 189, 45-62, 10.1016/j.pharmthera.2018.04.004.
- Hiroyuki Yamamoto; Kohzoh Imai; Microsatellite instability: an update. Archives of Toxicology 2015, 89, 899-921, 10.1007/s00204-015-1474-0.
- Jinru Shia; Immunohistochemistry versus Microsatellite Instability Testing For Screening Colorectal Cancer Patients at Risk For Hereditary Nonpolyposis Colorectal Cancer Syndrome. The Journal of Molecular Diagnostics 2008, 10, 293-300, 10.2353/jmoldx.2008.080031.
- Valerie Lee; Adrian Murphy; Dung T. Le; Luis A. Diaz; Mismatch Repair Deficiency and Response to Immune Checkpoint Blockade. The Oncologist 2016, 21, 1200-1211, 10.1634/theoncologist.2016-0046.
- Alexandre Luiz Affonso Fonseca; Fabio C. Prosdocimi; Bianca Bianco; Matheus Moreira Perez; Fernando Luiz Affonso Fonseca; Beatriz Da Costa Aguiar Alves Reis; Involvement of repair genes in oral cancer: A systematic review. Cell Biochemistry and Function 2019, 37, 572-577, 10.1002/cbf.3428.
- Shiladitya Sengupta; Susmita Chakrabarti; Anup Roy; Chinmay K. Panda; Susanta Roychoudhury; Inactivation of human mutL homolog 1 and mutS homolog 2 genes in head and neck squamous cell carcinoma tumors and leukoplakia samples by promoter hypermethylation and its relation with microsatellite instability phenotype. Cancer 2007, 109, 703-712, 10.1002/cncr.22430.
- Manel Esteller; Epigenetics in Cancer. New England Journal of Medicine 2008, 358, 1148-1159, 10.1056/nejmra072067.
- Patrícia Carlos Caldeira; Mauro Henrique Nogueira Guimarães Abreu; Aline C. Batista; Maria Auxiliadora Vieira Do Carmo; hMLH1 immunoexpression is related to the degree of epithelial dysplasia in oral leukoplakia. Journal of Oral Pathology and Medicine 2010, 40, 153-159, 10.1111/j.1600-0714.2010.00963.x.
- Patrícia Carlos Caldeira; Maria Cássia Ferreira Aguiar; Ricardo Alves Mesquita; Maria Auxiliadora Vieira Do Carmo; Oral leukoplakias with different degrees of dysplasia: comparative study of hMLH1, p53, and AgNOR. Journal of Oral Pathology and Medicine 2011, 40, 305-311, 10.1111/j.1600-0714.2010.01000.x.
- Pinaki Mondal; Sayantan Datta; Guru Prasad Maiti; Aradhita Baral; Ganga Nath Jha; Chinmay Kumar Panda; Shantanu Chowdhury; Saurabh Ghosh; Bidyut Roy; Susanta Roychoudhury; et al. Comprehensive SNP Scan of DNA Repair and DNA Damage Response Genes Reveal Multiple Susceptibility Loci Conferring Risk to Tobacco Associated Leukoplakia and Oral Cancer. PLOS ONE 2013, 8, e56952, 10.1371/journal.pone.0056952.
- Denise Hélen Imaculada Pereira De Oliveira; Maria Luiza Diniz De Sousa Lopes; Dmitry José De Santana Sarmento; Lélia Maria Guedes Queiroz; Márcia Cristina Da Costa Miguel; Éricka Janine Dantas Da Silveira; Relationship between the epithelial expression of hMLH1, MDM2, and p63 and lower lip carcinogenesis. Journal of Oral Pathology and Medicine 2013, 43, 357-363, 10.1111/jop.12144.
- Patrick K. Ha; Thomas A. Pilkington; William H. Westra; James Sciubba; David Sidransky; Joseph A. Califano; Progression of microsatellite instability from premalignant lesions to tumors of the head and neck. International Journal of Cancer 2002, 102, 615-617, 10.1002/ijc.10748.
- S Sanguansin; S Petmitr; P Punyarit; V Vorasubin; W Weerapradist; R Surarit; HMSH2 gene alterations associated with recurrence of oral squamous cell carcinoma.. Journal of Experimental & Clinical Cancer Research 2006, 25, 251-257, .
- R Czerninski; S Krichevsky; Y Ashhab; D Gazit; V Patel; D Ben-Yehuda; Promoter hypermethylation of mismatch repair genes,hMLH1andhMSH2in oral squamous cell carcinoma. Oral Diseases 2009, 15, 206-213, 10.1111/j.1601-0825.2008.01510.x.
- M.J. Julia Ashazila; T P Kannan; R.N. Venkatesh; B.P. Hoh; Microsatellite instability and loss of heterozygosity in oral squamous cell carcinoma in Malaysian population. Oral Oncology 2011, 47, 358-364, 10.1016/j.oraloncology.2011.03.005.
- I. González-Ramírez; V. Ramirez-Amador; Maria Esther Irigoyen-Camacho; Y. Sánchez-Pérez; G. Anaya-Saavedra; M. Granados-García; F. García-Vázquez; C.M. García-Cuellar; hMLH1 promoter methylation is an early event in oral cancer. Oral Oncology 2011, 47, 22-26, 10.1016/j.oraloncology.2010.10.002.
- Vo Thi Thuong Lan; Nguyen Thu Trang; Doan Thi Hong Van; Trang Nguyen Thu; Ta Van To; Vuong Dieu Linh; Nguyen Quynh Uyen; A methylation-specific dot blot assay for improving specificity and sensitivity of methylation-specific PCR on DNA methylation analysis. International Journal of Clinical Oncology 2015, 20, 839-845, 10.1007/s10147-014-0780-5.
- Thanaa Ea Helal; Mona T. Fadel; Abdalla K. El-Thobbani; Amira M. El-Sarhi; Immunoexpression of p53 and hMSH2 in oral squamous cell carcinoma and oral dysplastic lesions in Yemen: Relationship to oral risk habits and prognostic factors. Oral Oncology 2012, 48, 120-124, 10.1016/j.oraloncology.2011.08.024.
- Maryam Jessri; Andrew J Dalley; Camile S. Farah; hMSH6: a potential diagnostic marker for oral carcinoma in situ. Journal of Clinical Pathology 2014, 68, 86-90, 10.1136/jclinpath-2014-202411.
- Guilherme Augusto Silva Nogueira; Gustavo Jacob Lourenço; Camila Borges Martins Oliveira; Fernando Augusto Lima Marson; Leisa Lopes-Aguiar; Ericka Francislaine Dias Costa; Tathiane Regine Penna Lima; Vitor Teixeira Liutti; Frederico Leal; Vivian Castro Antunes Santos; et al. Association between genetic polymorphisms in DNA mismatch repair-related genes with risk and prognosis of head and neck squamous cell carcinoma. International Journal of Cancer 2015, 137, 810-818, 10.1002/ijc.29435.
- Ritu Jha; Poonam Gaur; S.C. Sharma; Satya N. Das; Single nucleotide polymorphism in hMLH1 promoter and risk of tobacco-related oral carcinoma in high-risk Asian Indians. Gene 2013, 526, 223-227, 10.1016/j.gene.2013.05.014.
- Nicola Whiffin; Peter Broderick; Steven J. Lubbe; Andrea L. Pittman; Steven Penegar; Ian Chandler; Richard S Houlston; MLH1-93G > A is a risk factor for MSI colorectal cancer. Carcinogenesis 2011, 32, 1157-1161, 10.1093/carcin/bgr089.
- P. Slováková; L. Majerová; T. Matakova; M. Skerenova; E. Kavcová; E. Halasova; Mismatch Repair Gene Polymorphisms and Association with Lung Cancer Development. Taurine 6 2014, 833, 15-22, 10.1007/5584_2014_83.
- Tomasz Popławski; A Sobczuk; J Sarnik; E Pawlowska; Janusz Blasiak; POLYMORPHISM OF DNA MISMATCH REPAIR GENES IN ENDOMETRIAL CANCER. Experimental Oncology 2015, 37, 44-7, 10.31768/2312-8852.2015.37(1):44-47.
- Stamatios Theocharis; Jerzy Klijanienko; Constantinos Giaginis; Jose Rodriguez; Thomas Jouffroy; Angelique Girod; Daniel Point; Gerasimos Tsourouflis; Xavier Sastre-Garau; Expression of DNA repair proteins, MSH2, MLH1 and MGMT in mobile tongue squamous cell carcinoma: associations with clinicopathological parameters and patients’ survival. Journal of Oral Pathology and Medicine 2010, 40, 218-226, 10.1111/j.1600-0714.2010.00945.x.
- Kartik Vasan; Laveniya Satgunaseelan; Sunaina Anand; Rebecca Asher; Christina Selinger; Tsu-Hui (Hubert) Low; Carsten E. Palme; Jonathan R. Clark; Ruta Gupta; Tumour mismatch repair protein loss is associated with advanced stage in oral cavity squamous cell carcinoma.. Pathology 2019, 51, 688-695, 10.1016/j.pathol.2019.08.005.
- Sonia Gupta; Manveen Kaur Jawanda; Oral lichen planus: An update on etiology, pathogenesis, clinical presentation, diagnosis and management. Indian Journal of Dermatology 2015, 60, 222-9, 10.4103/0019-5154.156315.
- Changchang Li; Xiaoqiong Tang; Xiaoyan Zheng; Shuqi Ge; Hao Wen; Xiaoqiong Lin; Zhiwei Chen; Liming Lu; Global Prevalence and Incidence Estimates of Oral Lichen Planus. JAMA Dermatology 2020, 156, 172, 10.1001/jamadermatol.2019.3797.
- Flavio Juliano Garcia Santos Pimenta; Expression of hMSH2 protein of the human DNA mismatch repair system in oral lichen planus. International Journal of Medical Sciences 2004, 1(3), 146-151, 10.7150/ijms.1.146.
- Ludmilla R. Souza; Thiago Fonseca-Silva; Camila S. Pereira; Erivelton P. Santos; Lucianne C. Lima; Heloisa A. Carvalho; Ricardo S. Gomez; Andre L. S. Guimaraes; Alfredo M. B. De Paula; Immunohistochemical analysis of p53, APE1, hMSH2 and ERCC1 proteins in actinic cheilitis and lip squamous cell carcinoma. Histopathology 2011, 58, 352-360, 10.1111/j.1365-2559.2011.03756.x.
- Dmitry José De Santana Sarmento; Wagner Leite De Almeida; Lélia Maria Guedes Queiroz; Gustavo Pina Godoy; Maria Carmen Fontoura Nogueira Da Cruz; Éricka Janine Dantas Da Silveira; Márcia Cristina Da Costa Miguel; Immunohistochemical analysis of mismatch proteins in carcinogenesis of the lower lip. Histopathology 2013, 63, 371-377, 10.1111/his.12197.
- Maria Luiza Diniz De Sousa Lopes; Denise Hélen Imaculada Pereira De Oliveira; Dmitry José De Santana Sarmento; Lélia Maria Guedes Queiroz; Márcia Cristina Da Costa Miguel; Éricka Janine Dantas Da Silveira; Correlation between cell cycle proteins and hMSH2 in actinic cheilitis and lip cancer. Archives of Dermatological Research 2016, 308, 165-171, 10.1007/s00403-016-1625-z.
- Zubeyde Yalniz; Semra Demokan; Yusufhan Suoglu; Murat Ulusan; Nejat Dalay; Assessment of microsatellite instability in head and neck cancer using consensus markers. Molecular Biology Reports 2010, 37, 3541-3545, 10.1007/s11033-010-0001-x.
- S. Demokan; Y. Suoglu; D. Demir; M. Gozeler; Nejat Dalay; Microsatellite instability and methylation of the DNA mismatch repair genes in head and neck cancer. Annals of Oncology 2006, 17, 995-999, 10.1093/annonc/mdl048.
- Eveson, J.W.; Yeudall, A. What is the evidence for the progression from benign to malignant pleomorphic adenoma? In Controversies in the Management of Salivary Gland Disease; McGurk, M., Renehan, A., Eds.; Oxford University Press: Oxford, UK, 2001; pp. 105–113.
- Iacopo Sardi; Alessandro Franchi; Gennaro Ferriero; Aimo Frittelli; Luca Bruschini; Enrico Montali; Oreste Gallo; Prediction of recurrence by microsatellite analysis in head and neck cancer. Genes, Chromosomes and Cancer 2000, 29, 201-206, 10.1002/1098-2264(2000)9999:9999<::aid-gcc1031>3.0.co;2-x.
- P M Speight; A W Barrett; Salivary gland tumours. Oral Diseases 2002, 8, 229-240, 10.1034/j.1601-0825.2002.02870.x.
- S.I. Tobón-Arroyave; Gloria Amparo Flórez-Moreno; Juan Fernando Jaramillo-Cárdenas; Juan David Arango-Uribe; Diana María Isaza-Guzmán; Javier Rendón-Henao; Expression of hMLH1 and hMSH2 proteins in pleomorphic adenoma of minor salivary glands: Relationship with clinical and histologic findings. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology 2009, 108, 227-236, 10.1016/j.tripleo.2009.02.033.
- Harlinde De Schutter; Marijke Spaepen; Sofie Van Opstal; Vincent Vander Poorten; Erik Verbeken; Sandra Nuyts; The prevalence of microsatellite instability in head and neck squamous cell carcinoma. Journal of Cancer Research and Clinical Oncology 2008, 135, 485-490, 10.1007/s00432-008-0476-1.
- K. Ohki; H. Kumamoto; R. Ichinohasama; M. Suzuki; T. Yamaguchi; S. Echigo; K. Motegi; K. Ooya; Genetic analysis of DNA microsatellite loci in salivary gland tumours: comparison with immunohistochemical detection of hMSH2 and p53 proteins. International Journal of Oral and Maxillofacial Surgery 2001, 30, 538-544, 10.1054/ijom.2001.0161.
- Graziella Castrilli; Alfredo Fabiano; Giampiero La Torre; Luca Marigo; Chiara Piantelli; Giorgio Perfetti; Franco O. Ranelletti; Mauro Piantelli; Expression of hMSH2 and hMLH1 proteins of the human DNA mismatch repair system in salivary gland tumors. Journal of Oral Pathology and Medicine 2002, 31, 234-238, 10.1034/j.1600-0714.2002.310407.x.
- Yimin Wang; Jonathan Irish; Christina Macmillan; Dale Brown; Yali Xuan; Curtiss Boyington; Patrick Gullane; Suzanne Kamel-Reid; High frequency of microsatellite instability in young patients with head-and-neck squamous-cell carcinoma: Lack of involvement of the mismatch repair geneshMLH1 ANDhMSH2. International Journal of Cancer 2001, 93, 353-360, 10.1002/ijc.1337.
- Stephane Temam; Odile Casiraghi; Jean-Baptiste Lahaye; Jacques Bosq; Xian Zhou; Morbize Julieron; Gérard Mamelle; J. Jack Lee; Li Mao; Bernard Luboinski; et al. Tetranucleotide Microsatellite Instability in Surgical Margins for Prediction of Local Recurrence of Head and Neck Squamous Cell Carcinoma. Clinical Cancer Research 2004, 10, 4022-4028, 10.1158/1078-0432.ccr-04-0199.
- Jin-Ching Lin; Chen-Chi Wang; Rong-San Jiang; Wen-Yi Wang; Shih-An Liu; Impact of microsatellite alteration in surgical margins on local recurrence in oral cavity cancer patients. European Archives of Oto-Rhino-Laryngology 2016, 274, 431-439, 10.1007/s00405-016-4215-y.
- Shih-An Liu; C. C. Wang; R. S. Jiang; W. Y. Wang; J. C. Lin; Genetic analysis of surgical margins in oral cavity cancer. British Journal of Surgery 2018, 105, e142-e149, 10.1002/bjs.10693.
- Danely P. Slaughter; Harry W. Southwick; Walter Smejkal; “Field cancerization” in oral stratified squamous epithelium. Clinical implications of multicentric origin. Cancer 1953, 6, 963-968, 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q.
- C.R. Leemans; Boudewijn J. M. Braakhuis; Ruud H. Brakenhoff; The molecular biology of head and neck cancer. Nature Reviews Cancer 2010, 11, 9-22, 10.1038/nrc2982.
- Erich M. Sturgis; Peng Wei; Genetic susceptibility–molecular epidemiology of head and neck cancer. Current Opinion in Oncology 2002, 14, 310-317, 10.1097/00001622-200205000-00010.
- Wei-Long Zhang; Zhuo-Li Zhu; Mei-Chang Huang; Ya-Jie Tang; Xin-Hua Liang; Ya-Ling Tang; Susceptibility of Multiple Primary Cancers in Patients With Head and Neck Cancer: Nature or Nurture?. Frontiers in Oncology 2019, 9, 1275, 10.3389/fonc.2019.01275.
- S. Piccinin; Daniela Gasparotto; T. Vukosavljevic; L. Barzan; S. Sulfaro; Roberta Maestro; M. Boiocchi; Microsatellite instability in squamous cell carcinomas of the head and neck related to field cancerization phenomena.. British Journal of Cancer 1998, 78, 1147-1151, 10.1038/bjc.1998.644.
- D Ronchetti; E Arisi; A Neri; G Pruneri; B Digiuni; G Sambataro; O Gallo; L Pignataro; Microsatellite analyses of recurrence or second primary tumor in head and neck cancer.. Anticancer Research 2005, 25, 2771-5, .
- Alberto Deganello Md; Gianni Gitti; Giuditta Mannelli; Giuseppe Meccariello; Oreste Gallo; Risk Factors for Multiple Malignancies in the Head and Neck. Otolaryngology–Head and Neck Surgery 2013, 149, 105-111, 10.1177/0194599813484273.
- Paolo Manca; Luis E. Raez; Matthew Salzberg; Jorge Sanchez; Brian Hunis; Christian Rolfo; The value of immunotherapy in head and neck cancer. Expert Opinion on Biological Therapy 2018, 19, 35-43, 10.1080/14712598.2019.1556637.
- M. Adachi; Kei Ijichi; Yasuhisa Hasegawa; Hideaki Nakamura; Tetsuya Ogawa; Nobutake Kanematsu; Human MLH1 status can potentially predict cisplatin sensitivity but not microsatellite instability in head and neck squamous cell carcinoma cells. Experimental and Therapeutic Medicine 2010, 1, 93-96, 10.3892/etm_00000017.
- Arnauld Cabelguenne; Ollivier Laccourreye; Isabelle De Waziers; Richard Hamelin; Daniel Brasnu; Philippe Beaune; Pierre Laurent-Puig; Microsatellite analysis and response to chemotherapy in head-and-neck squamous-cell carcinoma. International Journal of Cancer 1999, 84, 410-415, 10.1002/(sici)1097-0215(19990820)84:4<410::aid-ijc14>3.0.co;2-j.
- Efterpi Papouli; Petr Cejka; Josef Jiricny; Dependence of the Cytotoxicity of DNA-Damaging Agents on the Mismatch Repair Status of Human Cells. Cancer Research 2004, 64, 3391-3394, 10.1158/0008-5472.CAN-04-0513.
- P. Karran; Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis 2001, 22, 1931-1937, 10.1093/carcin/22.12.1931.
- Wei Yang; Ken-Wing Lee; Raghvendra M. Srivastava; Fengshen Kuo; Chirag Krishna; Diego Chowell; Vladimir Makarov; Douglas Hoen; Martin G. Dalin; Leonard Wexler; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nature Medicine 2019, 25, 767-775, 10.1038/s41591-019-0434-2.
- Rachel Rosenthal; The TRACERx consortium; Elizabeth Larose Cadieux; Roberto Salgado; Maise Al Bakir; David A. Moore; Crispin T. Hiley; Tom Lund; Miljana Tanić; James L. Reading; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature Cell Biology 2019, 567, 479-485, 10.1038/s41586-019-1032-7.
- Xiaomo Wu; Zhongkai Gu; Yang Chen; Borui Chen; Wei Chen; Liqiang Weng; Zuwu Wei; Application of PD-1 Blockade in Cancer Immunotherapy. Computational and Structural Biotechnology Journal 2019, 17, 661-674, 10.1016/j.csbj.2019.03.006.
- Yoshiko Iwai; Masayoshi Ishida; Yoshimasa Tanaka; Taku Okazaki; Tasuku Honjo; Nagahiro Minato; Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proceedings of the National Academy of Sciences 2002, 99, 12293-12297, 10.1073/pnas.192461099.
- Ryan B. Corcoran; Axel Grothey; Efficacy of Immunotherapy in Microsatellite-Stable or Mismatch Repair Proficient Colorectal Cancer—Fact or Fiction?. JAMA Oncology 2020, 6, 823, 10.1001/jamaoncol.2020.0504.
- Laetitia Nebot-Bral; Clelia Coutzac; Patricia L. Kannouche; Nathalie Chaput; Why is immunotherapy effective (or not) in patients with MSI/MMRD tumors?. Bulletin du Cancer 2019, 106, 105-113, 10.1016/j.bulcan.2018.08.007.
- Hiro Sato; Penny Jeggo; Atsushi Shibata; Regulation of programmed death‐ligand 1 expression in response to DNA damage in cancer cells: Implications for precision medicine. Cancer Science 2019, 110, 3415-3423, 10.1111/cas.14197.
- Dung T. Le; Jennifer N. Uram; Hao Wang; Bjarne R. Bartlett; Holly Kemberling; Aleksandra D. Eyring; Andrew D. Skora; Brandon S. Luber; Nilofer S. Azad; Dan Laheru; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine 2015, 372, 2509-2520, 10.1056/nejmoa1500596.
- Rajarsi Mandal; Robert M. Samstein; Ken-Wing Lee; Jonathan J. Havel; Hao Wang; Chirag Krishna; Erich Y. Sabio; Vladimir Makarov; Fengshen Kuo; Pedro Blecua; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti–PD-1 immunotherapy response. Science 2019, 364, 485-491, 10.1126/science.aau0447.
- Khalil Saleh; Roland Eid; Fady Gh Haddad; Nadine Khalife-Saleh; Hampig R Kourie; New developments in the management of head and neck cancer – impact of pembrolizumab. Therapeutics and Clinical Risk Management 2018, 14, 295-303, 10.2147/tcrm.s125059.
- Robert L. Ferris; George Blumenschein; Jerome Fayette; Joel Guigay; A. Dimitrios Colevas; Lisa Licitra; Kevin J. Harrington; Stefan Kasper; Everett E. Vokes; Caroline Even; et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncology 2018, 81, 45-51, 10.1016/j.oraloncology.2018.04.008.
- Tanguy Y. Seiwert; Barbara Burtness; Ranee Mehra; Jared Weiss; Raanan Berger; Joseph Paul Eder; Karl Heath; Terrill McClanahan; Jared Lunceford; Christine Gause; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. The Lancet Oncology 2016, 17, 956-965, 10.1016/s1470-2045(16)30066-3.
- Laura Q.M. Chow; Robert Haddad; Shilpa Gupta; Amit Mahipal; Ranee Mehra; Makoto Tahara; Raanan Berger; Joseph Paul Eder; Barbara Burtness; Se-Hoon Lee; et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. Journal of Clinical Oncology 2016, 34, 3838-3845, 10.1200/jco.2016.68.1478.
- PD-L1 IHC 22C3 pharmDx for Autostainer Link 48. . www.agilent.com. Retrieved 2020-12-26
- Jaimin Patel; Dylan A. Levy; Shaun A. Nguyen; Hannah M. Knochelmann Bs; Terry A. Day; Impact of PD‐L1 expression and human papillomavirus status in anti‐PD1/PDL1 immunotherapy for head and neck squamous cell carcinoma—Systematic review and meta‐analysis. Head & Neck 2020, 42, 774-786, 10.1002/hed.26036.
- Joshua Bauml; Tanguy Y. Seiwert; David G. Pfister; Francis Worden; Stephen V. Liu; Jill Gilbert; Nabil F. Saba; Jared Weiss; Lori Wirth; Ammar Sukari; et al. Pembrolizumab for Platinum- and Cetuximab-Refractory Head and Neck Cancer: Results From a Single-Arm, Phase II Study. Journal of Clinical Oncology 2017, 35, 1542-1549, 10.1200/jco.2016.70.1524.
- Magalie P. Tardy; Ilaria Di Mauro; Nathalie Ebran; Sadal Refae; Alexandre Bozec; Karen Benezery; Frédéric Peyrade; Joel Guigay; Anne Sudaka-Bahadoran; Cecile Badoual; et al. Microsatellite instability associated with durable complete response to PD-L1 inhibitor in head and neck squamous cell carcinoma. Oral Oncology 2018, 80, 104-107, 10.1016/j.oraloncology.2018.04.001.