Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Respiratory System
The tumor microenvironment plays a key role in the progression of lung tumorigenesis, progression, and metastasis. Recent data reveal that disseminated tumor cells (DTCs) appear to play a key role in the development and progression of lung neoplasiaby driving immune system dysfunction and established immunosuppression, which is vital for evading the host immune response.
- disseminated tumor cells
- immunosuppression
- lung cancer
- metastasis
1. Introduction
The metastatic spread of tumor cells is a key process for cancer progression. Accumulating evidence reveals that the spread of tumor cells can occur even at the early stages of carcinogenesis [1,2]. This process begins with the local invasion of primary tumor cells into surrounding healthy tissues: intravasation, extravasation formation of micrometastatic colonies, and the subsequent proliferation of microscopic colonies into metastatic lesions, a process termed as colonization (Figure 1). These disseminated tumor cells (DTCs) can migrate to distant organs and initiate tumor regrowth, triggering cancer recurrence and subsequent metastasis. Recent studies demonstrate that DTC-related tumor heterogeneity inside the tumor microenvironment (TME) plays a crucial role in tumor growth and expansion. The clinical significance of DTCs is proven by many studies, indicating their decisive and vital role in chemotherapy/immunotherapy resistance, tumor, recurrence, and metastasis [3,4]. In this review, we will investigate and analyze the various signaling pathways used by DTCs and their crosstalk with immune or stromal cells inside the TME, and we will also explore the development of new approaches and therapies that are specifically designed to target DTC-related metastasis.
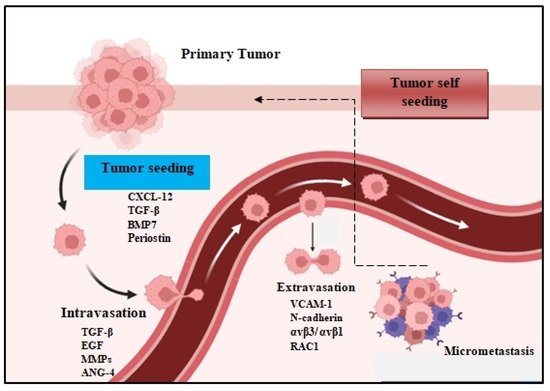
Figure 1. The metastatic colonization cascade. The various steps of metastasis are depicted: intravasation, circulation, extravasation, and colonization of distant organs. In many tumors, cancer cells can return to the primary tumor (tumor self seeding) and accelerate its expansion.
2. Early Cancer Cell Dissemination
The mechanism of dissemination alters the phenotypic morphology of tumor cells in order to obtain traits that allow them to leave the primary tumor and spread (metastasize) to distant tissues or organs via blood circulation [5]. These circulating tumor cells (CTCs) can extravasate into secondary tissues, where they develop into DTCs. Active dissemination can occur through the transformation of epithelial cells into a more mesenchymal phenotype through an altered EMT process [6]. In many cases tumor cells disseminate in situ even before the detection of the primary tumor. This aspect of early dissemination, where DTCs develop in parallel with the primary tumor, is highly significant to understanding the unique molecular biology of metastatic colonization [2,7]. Recently, sustained lung inflammation was shown to convert disseminated, dormant cancer cells into aggressively growing metastases. This sustained inflammation induced the formation of neutrophil extracellular traps (NETs), and these were required for the awakening of dormant tumor cells [8]. In general, the TME is the critical regulator of cancer cell dissemination and cancer progression. This immunosuppressive and hypoxic TME can transform tumor cells in a prolonged quiescent state into active DTCs and in due course mediate metastatic outgrowth in distant organs or tumor recurrence [9,10].
2.1. Tumor Cell Dissemination
The peripheral blood contains numerous sources of tumor-derived material, like circulating tumor DNA (ctDNA), CTCs, and exosomes, which support tumorigenesis. Furthermore, the extracellular matrix (ECM) plays a critical role in the maintenance of the tumorigenic profile of DTCs [11,12]. These factors determine both the survival rate and adaptation of DTCs in metastatic sites. Apart from establishing metastatic hotspots, DTCs often come back to the primary tumor to accelerate their development [13]. This hypothesis of tumor self-feeding has broadened our knowledge of the pathogenesis of metastasis [14]. Furthermore, endothelial cells, which form the blood vessels that support the TME by providing nutrients and oxygen, also play critical roles in metastatic dissemination. Specifically, the tumoral activation of the TLR3-SLIT2 axis in endothelium drives metastasis. The endothelial SLIT2 protein and its receptor ROBO1 promoted the migration of cancer cells towards endothelial cells and intravasation. Deleting endothelial Slit2 suppressed metastatic dissemination in mouse models of breast and lung cancer. Conversely, the deletion of tumoral Slit2 enhanced metastatic progression [4]. Similarly, tumor-mesenchymal stem cells (T-MSCs) as heterogeneous stromal cells promote lung cancer metastasis by inducing the expression of genes associated with an aggressive phenotype in primary lung cancer cells [15]. Apart from that, immune cells can also promote the initial metastatic dissemination of carcinoma cells from primary tumors. For example, CD11b(+)/Ly6G(+) neutrophils enhance metastasis formation via the inhibition of the natural killer cell function, which leads to a significant increase in intraluminal survival along with the extravasation/dissemination of tumor cells through the secretion of IL1β and matrix metalloproteinases [16].
2.2. Epithelial-Mesenchymal Plasticity
Both the epithelial to mesenchymal transition (EMT) and mesenchymal to epithelial transition (MET) are developmental programs that are activated during embryogenesis and tissue repair [17]. During carcinogenesis, these programs are being hijacked by tumor cells in order to increase their resistance to chemotherapy, acquire the cancer stem cell (CSC) phenotype, and amplify their metastatic dissemination potential [18,19] (Figure 2). DTCs can manipulate EMT signaling in order to promote their dissociation from the primary tumor and disseminate into blood circulation [20]. Because EMT/MET are highly dynamic and plastic, DTC employ several EMT-inducing transcription factors such as Twist, Snail, Slug, and Zeb1, which activate these mechanisms and prompt metastatic colonization [21]. The activation of EMT can also occur via epidermal growth factor (EGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), insulin growth factor (IGF), and PDGF. In many cases, DTCs cells transit through a series of EMT/MET states also named partial-EMT, where tumor cells possess both epithelial and newly acquired mesenchymal characteristics and display a high degree of phenotypic plasticity [22,23].
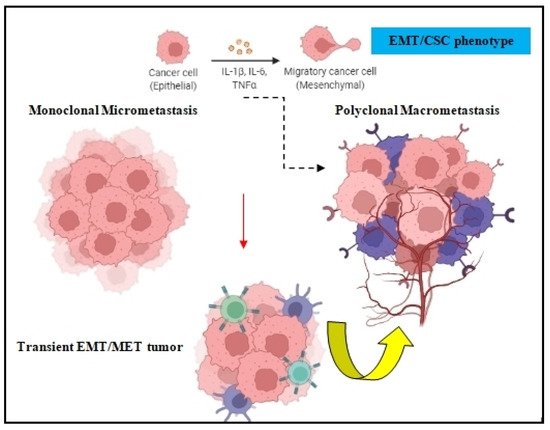
Figure 2. EMT in tumor cell dissemination. The EMT/CSC phenotype of DTCs can prompt monoclonal micrometastasis seeding to develop polyclonal macrometastasis, depending on the clonal and tumor-stromal cells interactions in the target organ.
3. Integrin Signaling
Integrins are transmembrane proteins that mediate cell adhesion, regulate the cell cycle, and organize the intracellular cytoskeleton [24]. Accumulating evidence reveals that tumor cells alter integrin signaling inside the TME, thus contributing to immunosuppression and immunoresistance due to their ability to control immune cell adhesion to endothelial cell layers followed by their trans-migration [25]. They are also involved in tumor expansion and progression by mediating angiogenesis, lymphangiogenesis, desmoplasia, and inflammation [26,27]. Tumor cell expression of the integrins αvβ3, αvβ5, a5β1, a6β4, a4β1, and αvβ6 is often associated with increased chemoresistance, disease progression, and poor survival [28,29].
Metastatic Colonization
Recent data reveal that in the early stages of the metastatic cascade, bidirectional communication between malignant cells and the TME is essential for maintaining tissue homeostasis and tumor expansion. Metastatic tumorigenesis is a heterogeneous disease progressing in a multistep process involving aberrant mutated tumor cells, altered phenotype immune cells, and highly polarized stromal cells. As a whole, metastatic colonization is a very ineffective mechanism, where only 0.01% of tumor cells survive to form micro-metastases. During this early phase of metastasis, cytoskeleton rearrangements within tumor cells in cooperation with aberrant differentiation signaling of Notch, WNT, and Hedgehog pathways guide tumor cell invasion. This metastatic cascade occurs via single tumor cells or cell clusters. Recently, the ability of CTCs to form clusters has been linked to increased metastatic potential. In particular, CTC clustering shapes DNA methylation to enable metastasis seeding and the promotion of stemness [30]. These CTC clusters can derive from multicellular groupings of primary tumor cells held together with intercellular adhesion, and they greatly contribute to the metastatic spread of cancer [31]. Most CTC clusters are heterogeneous and adopt multiple ways to enhance their metastatic potential, including homotypic clustering and heterotypic interactions with immune and stromal cells [32,33].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14153626
This entry is offline, you can click here to edit this entry!