Methylmercury (MeHg) is a well-known neurotoxicant that causes severe intoxication in humans. Research progress has pointed out the importance of oxidative stress in the pathogenesis of MeHg toxicity. MeHg-induced intracellular relative selenium deficiency due to the greater affinity of MeHg for selenohydryl groups and selenides leads to failure in the recoding of a UGA codon for selenocysteine and results in the degradation of antioxidant selenoenzyme mRNA by nonsense-mediated mRNA decay. The defect of antioxidant selenoenzyme replenishment exacerbates MeHg-mediated oxidative stress.
- methylmercury
- oxidative stress
- antioxidant selenoenzymes
- binding affinity
- selenium deficiency
- selenocysteine
- premature termination codon
- nonsense-mediated mRNA decay
- posttranscriptional defect
Methylmercury (MeHg) is a well-established neurotoxicant that affects various cellular functions depending on the cellular context and developmental phase. Research progress has pointed out the importance of oxidative stress in the pathogenesis of MeHg toxicity. MeHg has a high affinity for selenohydryl groups, sulfhydryl groups, and selenides [1]. It has been clarified that such affinity characteristics causes the impairment of antioxidant enzymes and proteins, resulting in the disruption of antioxidant systems.
MeHg-mediated increases in intracellular reactive oxygen species (ROS) causes changes in antioxidant gene expression. Our previous study demonstrated that MeHg exposure upregulated manganese (Mn)-SOD, copper, zinc (Cu, Zn)-SOD, catalase, and thioredoxin reductase 1 (TrxR1) mRNAs [2]. The upregulation of these mRNAs was mediated by ROS because treatment with the antioxidant Trolox suppressed the increase in these mRNAs. In contrast, selenoenzyme glutathione peroxidase 1 (GPx1) mRNA was downregulated despite its decreased activity in vitro and in vivo [2]. In addition, Trolox failed to rescue such GPx1 mRNA decrease. Our in situ antioxidative enzymes expression analyses using laser micro-dissected mouse cerebrocortical neuron samples also revealed downregulation of GPx1 mRNA [3]. This is intriguing because oxidative stress due to the general burden of H2O2 caused upregulation of GPx1 mRNA, indicating that the MeHg-induced GPx1 mRNA decrease is specific to the burden of MeHg [2].
GPx1 has a single selenocysteine (Sec), in which selenium (Se) is co-translationally inserted. Sec is encoded by a UGA codon, which shares a common codon to function as a terminator for protein synthesis. The biosynthesis of Sec occurs on its tRNA (Sec tRNA [Ser]Sec), unlike the other 20 amino acids. Once activated Se is donated to the structure, Sec is completed [4]. The insertion of Sec into protein requires the Sec insertion sequence (SECIS) [5], SECIS binding protein (SBP2) [6], and Sec-specific elongation factor [7, 8]. Under Se deficiency, however, the UGA codon for Sec may be recognized as a nonsense codon, known as a premature termination codon (PTC), due to the incomplete biosynthesis of Sec. mRNAs harboring PTCs are known to be deleted by nonsense-mediated mRNA decay (NMD), an mRNA quality control mechanism that is executed when PTC is located sufficiently upstream of the exon–exon junction [9-12]. A previous report demonstrated that Sec on GPx1 mRNA that resides 105 nucleotides upstream of the sole exon–exon junction was recognized as a PTC and degraded by NMD under active Se-deficient conditions [13].
It is known that the selenohydryl group has a high affinity for mercury (Hg) compared to those of the sulfhydryl and amino groups. The order of binding affinity of the coordination groups toward MeHg is as follows: SeH > SH ≥ Se-Se > NH2 > S-S [1]. The high affinity of MeHg for the selenohydryl group and selenide should cause relative intracellular Se-deficient conditions under MeHg exposure. Our previous study demonstrated that the MeHg-induced decrease in GPx1 mRNA is a post-transcriptional event by NMD, enhanced degradation of mRNA that is most likely mediated by cellular Se deficiency [2]. This finding was confirmed by two studies: (1) MeHg-induced decrease in GPx1 mRNA was rescued by pretreatment with sodium selenite, and (2) MeHg-induced decrease in GPx1 mRNA was inhibited in siRNA-mediated NMD component knockdown cells. In contrast to GPx1, mRNA of another antioxidant selenoenzyme, TrxR1, was not downregulated by MeHg exposure. The Sec codon UGA-498 on TrxR1 that resides in the last exon cannot be a substrate for NMD because at least one downstream intron is required to trigger NMD [14-16]. In theory, the TrxR1 protein synthesized by NMD-skipped TrxR1 mRNA should be truncated because the Sec codon is recognized as a nonsense codon under Se-deficient conditions. The different pathways involved in the synthesis of GPx1 and TrxR1 under the sufficient or MeHg-induced deficient active form of Se are summarized in Figure 1.
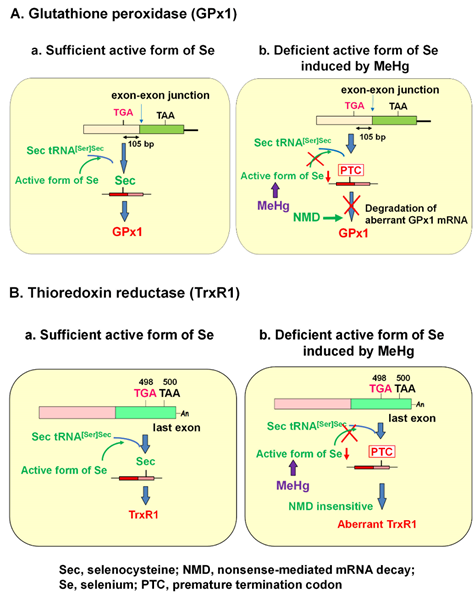
Figure 1. Posttranscriptional effect of methylmercury (MeHg) on antioxidant selenoenzymes. (A) Glutathione peroxidase 1 (GPx1). The encoded UGA codon for selenocysteine (Sec) resides 105 nucleotides upstream of the sole exon–exon junction. When a UGA codon is recognized as a Sec codon under sufficient active form of selenium (Se) (left panel), GPx1 is synthesized. However, since UGA codon is recognized as a nonsense codon under MeHg-induced active Se deficiency (right), GPx1 mRNA should be a natural substrate for nonsense-mediated mRNA decay (NMD; right panel). (B) Thioredoxin reductase 1 (TrxR1). The Sec codon UGA-498 resides in the last exon on TrxR1 mRNA; thus, TrxR1 mRNA cannot be a substrate for NMD even when a UGA codon is recognized as a nonsense codon under MeHg-induced Se deficiency and aberrant Trx1 is synthesized (right panel).
References
- Sugiura, Y.; Tamai, Y.; Tanaka, H., Selenium protection against mercury toxicity: high binding affinity of methylmercury by selenium-containing ligands in comparison with sulfur-containing ligands. Bioinorg Chem 1978, 9, (2), 167-80.
- Usuki, F.; Yamashita, A.; Fujimura, M., Post-transcriptional defects of antioxidant selenoenzymes cause oxidative stress under methylmercury exposure. The Journal of biological chemistry 2011, 286, (8), 6641-9.
- Fujimura, M.; Usuki, F., In situ different antioxidative systems contribute to the site-specific methylmercury neurotoxicity in mice. Toxicology 2017, 392, 55-63.
- Hatfield, D. L.; Gladyshev, V. N., How selenium has altered our understanding of the genetic code. Molecular and cellular biology 2002, 22, (11), 3565-76.
- Low, S. C.; Berry, M. J., Knowing when not to stop: selenocysteine incorporation in eukaryotes. Trends in biochemical sciences 1996, 21, (6), 203-8.
- Low, S. C.; Grundner-Culemann, E.; Harney, J. W.; Berry, M. J., SECIS-SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. The EMBO journal 2000, 19, (24), 6882-90.
- Fagegaltier, D.; Hubert, N.; Yamada, K.; Mizutani, T.; Carbon, P.; Krol, A., Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation. The EMBO journal 2000, 19, (17), 4796-805.
- Tujebajeva, R. M.; Copeland, P. R.; Xu, X. M.; Carlson, B. A.; Harney, J. W.; Driscoll, D. M.; Hatfield, D. L.; Berry, M. J., Decoding apparatus for eukaryotic selenocysteine insertion. EMBO reports 2000, 1, (2), 158-63.
- Frischmeyer, P. A.; Dietz, H. C., Nonsense-mediated mRNA decay in health and disease. Human molecular genetics 1999, 8, (10), 1893-900.
- Holbrook, J. A.; Neu-Yilik, G.; Hentze, M. W.; Kulozik, A. E., Nonsense-mediated decay approaches the clinic. Nature genetics 2004, 36, (8), 801-8.
- Maquat, L. E., Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nature reviews. Molecular cell biology 2004, 5, (2), 89-99.
- Yamashita, A.; Kashima, I.; Ohno, S., The role of SMG-1 in nonsense-mediated mRNA decay. Biochimica et biophysica acta 2005, 1754, (1-2), 305-15.
- Moriarty, P. M.; Reddy, C. C.; Maquat, L. E., Selenium deficiency reduces the abundance of mRNA for Se-dependent glutathione peroxidase 1 by a UGA-dependent mechanism likely to be nonsense codon-mediated decay of cytoplasmic mRNA. Molecular and cellular biology 1998, 18, (5), 2932-9.
- Nagy, E.; Maquat, L. E., A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends in biochemical sciences 1998, 23, (6), 198-9.
- Zhang, J.; Sun, X.; Qian, Y.; LaDuca, J. P.; Maquat, L. E., At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: a possible link between nuclear splicing and cytoplasmic translation. Molecular and cellular biology 1998, 18, (9), 5272-83.
- Sun, X.; Moriarty, P. M.; Maquat, L. E., Nonsense-mediated decay of glutathione peroxidase 1 mRNA in the cytoplasm depends on intron position. The EMBO journal 2000, 19, (17), 4734-44.
This entry is adapted from the peer-reviewed paper 10.3390/antiox9101004