Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Autophagy, a lysosome-mediated cellular degradation pathway, recycles intracellular components to maintain metabolic balance and survival. Autophagy plays an important role in tumor immunotherapy as a “double-edged sword” that can both promote and inhibit tumor progression.
- autophagy
- immunity
- cancer therapy
1. Introduction
Autophagy, first discovered by Belgian chemist Christine in 1963, is a process that is responsible for transporting damaged organelles, misfolded proteins and other macro molecules to lysosomes for degradation and regeneration [1]. It is a phenomenon that widely exists in eukaryotic cells. Several studies have shown that autophagy is triggered to varying degrees by processes such as angiogenesis and osteogenic differentiation during the differentiation of many cells [2,3,4]. Autophagy can be non-selective, when the cell is in an energy emergency to uptake generic cytoplasmic materials, or selective, to specifically remove damaged organelles, including mitochondria, ER, Golgi membranes and protein aggregates. The occurrence of autophagy mainly involves five major processes. In the autophagy induction phase, the counterbalanced control of mammalian target of rapamycin complex 1(mTORC1) and AMP-activated protein kinase (AMPK) via amino acid deprivation enhances ATG13 and unc-51-like autophagy-activating kinase 1 (ULK1) phosphorylation, and the ATG13–ATG1–ATG17 complex is formed. During vesicle nucleation, the lipid kinase activity of Vps34 facilitates the formation of a pre-autophagosomal structure (PAS), with Beclin 1 and ATG-14 like protein (ATG14L), inducing a ATG-conjugation cascade downstream. In the elongation stage of the autophagosome, the association of the ATG12–ATG5–ATG16 complex lapidates microtubule-associated protein light chain3 (LC3) or γ-aminobutyric acid receptor-associated protein (GABARAP) from the water-soluble form to a fat-soluble form. Lipidated LC3/GABARAP cooperates with other factors to elongate and close autophagosomes. WD repeat domain phosphoinositide-interacting protein 1 (WIPI 1) combines with Vps34-derived Ptdlns3phosphate (Ptdlns3P), cooperating with ATG2 and ATG9 to form autophagic organelles. During cargo assembling, LC3 II serves as a receptor for autophagy substrates, either to randomly capture or selectively target for degradation. For the final fusion with lysosomes, ultraviolet radiation resistance-associated gene protein (UVRAG) replaces ATG14L in the Vps34–Beclin 1 complex. The attachment of SNARE to the membrane of autophagosomes enables the fusion with lysosomes, following autolysosome degradation [5,6,7,8] (Figure 1a).
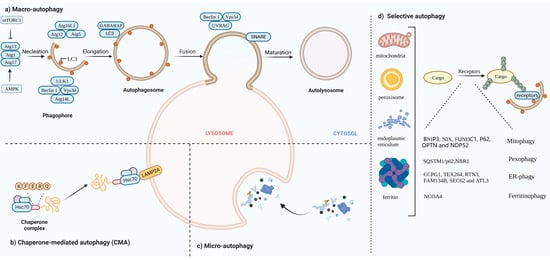
Figure 1. (a) Macro-autophagy: The occurrence of autophagy mainly involves five major processes: (1) Autophagy induction phase. In this phase, mammalian target of rapamycin complex 1(mTORC1) activity is inhibited; ATG13 phosphorylation is reduced; and the ATG13–ATG1–ATG17 complex is formed. (2) Vesicle nucleation. The Vps34–Beclin1 complex mediates the formation of pre-autophagosomal structure (PAS). (3) The elongation stage of the autophagosome. During this phase, the ATG12–ATG5–ATG16 complex is assembled and microtubule-associated protein light chain3 (LC3) LC3 is converted from the water soluble form (LC3 I) to a fat soluble form (LC3 II). (4) Random capturing or selective targeting for degradation. (5) Autophagosomes fuse with lysosomes and autophagosome cleavage. (b) CMA: Hsc70 specifically mediates protein degradation via receptor LAMP2A. (c) Micro-autophagy: Lysosomes directly engulf aggregates. (d) Selective autophagy: Specific intracellular components combine with individual cargo for autophagic degradation, including mitophagy, pexophagy, ER-phagy and ferritinophagy.
The tumor microenvironment (TME) is the surrounding environment in which the tumor grows and survives. It is now generally accepted that tumor cells, immune cells, stromal cells and the extracellular matrix (ECM) closely interact to form the main structure of the TME. These cells affect biological processes such as tumor growth and metastasis by secreting cytokines and releasing signaling molecules. Macrophages, lymphocytes, natural killer (NK) cells and dendritic cells (DCs), among the immune cells, are critical for tumor-cell killing and tumor control. However, some immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs) and type 2 polarized macrophages (M2 macrophages), are also present in the tumor microenvironment to counteract the anticancer immune response [9,10]. Tumor immunotherapy, such as oncolytic-virus therapies, cancer vaccines, cytokine therapies, adoptive cell transfer and immune checkpoint inhibitors targeting the TME, has made rapid progress in recent years. There is growing evidence that autophagy can be involved in the regulation of the innate and adaptive immunity [11]. At the same time, some immune cells and cytokines can also affect and regulate autophagy. Autophagy plays an important role in tumor immunotherapy as a “double-edged sword” that can both promote and inhibit tumor progression.
2. The Landscape and Forms of Autophagy
Autophagy can be divided into three categories based on how substances are packaged and transported, including macro-autophagy, micro-autophagy and chaperone-mediated autophagy (CMA) [12]. Macro-autophagy is the most common form of autophagy. Cytosolic substrates are wrapped by endoplasmic reticulum or mitochondria-derived bilayer membranes to form autophagosomes. After fusion with lysosomes, the cargo is degraded, and the resulting macromolecules are released back into the cytoplasmic matrix for reuse [13]. Micro-autophagy is the invagination of the lysosomal membrane itself, encapsulating and phagocytosing the substrates to be degraded in the cell and degrading them in a lysosome. The difference between micro-autophagy and macro-autophagy is the absence of autophagosomes [14,15]. Chaperone-mediated autophagy, independent of vesicle trafficking, often relies on chaperone protein Hsc70 to specifically degrade target proteins with a unique recognition pentapeptide motif (KFERQ-like motif) and receptor protein LAMP2A on the lysosomal membrane to recognize the binding-protein complex. The KFERQ group “guides” the target protein into the lysosome for degradation [16,17] (Figure 1b,c).
In this review, we mainly focus on macro-autophagy (hereafter referred to as autophagy). Generally, autophagy has been regarded as a non-selective transport of cytoplasmic components to lysosomes for bulk degradation since it appears to indiscriminately engulf cytosol. However, autophagy may also be highly selective. Transmission electron microscopy has detected autophagic compartments with different contents in mammalian cells, including mitochondria, ER and Golgi membranes [18]. Selective autophagy typically occurs under nutrient-rich conditions to remove damaged or redundant organelles such as mitochondria, endoplasmic reticulum (ER) and Golgi complexes [19]. Moreover, it depends on soluble or membrane-bound selective autophagy receptors (SARs) to degrade the specific intracellular components. According to autophagosomes with different contents, selective autophagy can be divided into mitophagy, lipophagy, ER-phagy, ferritinophagy, etc.
2.1. Mitophagy
In yeast and mammalian cells, damaged mitochondria can be selectively degraded via mitophagy, regulating the number of mitochondria in cells and maintaining normal function [20]. There are two main pathways that mediate mitophagy.
2.1.1. Receptor-Mediated Mitophagy
BNIP3, NIX, FUNDC1, PHB2. BNIP3, NIX and FUNDC1 receptors are localized to the OMM and directly interact with LC3 to mediate mitochondrial clearance [21,22,23]. NIX and Bnip3 promote the selective degradation of mitochondria during reticulocyte maturation [24,25]. The phosphorylation of BNIP3 and NIX enhances their interaction with LC3 [26]. After mitochondrial damaging, PHB2 and cardiolipin externalize to the OMM and interact with LC3 [27].
2.1.2. Ubiquitin-Mediated Mitophagy
PINK1/Parkin pathway: Under stress conditions, auto-phosphorylated PINK1 promotes Parkin recruitment and also leads to Parkin activation and the ubiquitination of substrates on damaged mitochondria that function as autophagy-mediated degradation signals [28,29]. P62, OPTN and NDP52 [30] recognize phosphorylated polyubiquitin chains on mitochondrial proteins and initiate autophagosome formation by binding to LC3.
2.2. Pexophagy
Under conditions of nutrient starvation or ROS burst, peroxisomes are degraded in an autophagic manner to maintain cellular homeostasis [31]. In response to ROS, ataxia-telangiectasia mutated (ATM) interacts with peroxisomal signal-receiving molecule PEX5, translocating to the peroxisome surface [32]. Then, phosphorylated PEX5 is further ubiquitinated by PEX2/10/12 and binds to p62/NBR1, interacting with LC3 to promote the occurrence of autophagy [33,34].
2.3. ER-Phagy
The ER is the largest organelle in the cell and has functions such as folding, processing and transporting proteins, and regulating cellular metabolism [35]. When unfolded proteins accumulate on the ER, ER-phagy is activated to degrade damaged ER, inhibit protein synthesis, relieve ER stress and enable cell survival. Six receptors have been identified in mammals that respond to the ER, CCPG1, TEX264, RTN3, FAM134B, SEC62 and ATL3, which contain at least one critical LIR/GIM domain interacting with LC3 II (or ATG8)/GABARAP to mediate the occurrence of ER autophagy [36,37,38,39,40,41,42].
2.4. Ferritinophagy
In the presence of low iron concentration in cells, ferritin is degraded in lysosomes through the activation of ferritinophagy. Iron (Fe) is stored in a ferritin complex containing ferritin heavy chain (FTH1) and light chain (FTL). Nuclear receptor coactivator 4 (NCOA4) binds to ferritin and mediates its delivery to the autophagosome. The fusion of autophagosomes with lysosomes results in ferritin degradation and subsequent iron release [43,44] (Figure 1d).
This entry is adapted from the peer-reviewed paper 10.3390/cells11152324
This entry is offline, you can click here to edit this entry!