Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Glioblastoma (GBM) is an aggressive primary brain tumor that is associated with a poor prognosis and quality of life. The standard of care has changed minimally over the past two decades and currently consists of surgery followed by radiotherapy (RT), concomitant and adjuvant temozolomide, and tumor treating fields (TTF). Factors such as tumor hypoxia and the presence of glioma stem cells contribute to the radioresistant nature of GBM. Small molecules and immunotherapy agents that have been studied in conjunction with RT in clinical trials are presented herein.
- glioblastoma
- radioresistance
- radiosensitizer
- glioma stem cell
- tumor hypoxia
1. Introduction
The rationale behind combining radiation and chemotherapy originates from the Steel paradigm [52]. Steel et al. proposed that synergy is driven by (1) spatial cooperation, (2) toxicity independence, (3) protection of normal tissues, and (4) enhancement of tumor response. The enhancement effect can be driven by inhibiting radiation-induced damage, reoxygenation following treatment, and/or improved drug access following RT.
Early studies demonstrated some chemotherapeutics such as cisplatin have the ability to sensitize tumor cells to RT, leading to greater radiation efficacy [53]. More recently, radiosensitizers have been developed that work through a variety of mechanisms: (1) Suppression of intracellular thiols or other radioprotective substances, (2) radiation-induced formation of cytotoxic substances via radiolysis of the sensitizer, (3) inhibition of the post-radiation cellular repair processes, (4) structural incorporation of thymine analogues into intracellular DNA, and (5) oxygen mimetic sensitizers [54,55].
Although other disease sites have found success with radiosensitizers, GBM has been particularly challenging due to its anatomic location (e.g., located beyond the blood–brain barrier), cell heterogeneity (e.g., cancer stem cells, tumor microtubes), and increased proliferation rate [56]. To date, TMZ is the most effective and widely used radiosensitizer in the treatment of GBM. TMZ increases the number of RT-induced double-strand DNA breaks as a result of a decrease in DNA repair capacity [57,58]. The content herein will focus on other small molecule and immunotherapy agents that have shown preclinical promise.
2. Pyrmidine Analogues
Gemcitabine is a difluoro-pyrimidine analog that is phosphorylated and incorporated into the DNA and RNA of cancer cells, leading to chain termination (Figure 2) [59]. The radiosensitizing effects of gemcitabine result from the depletion of phosphorylated deoxynucleotides and cell-cycle redistribution into the S-phase [60,61,62]. To date, gemcitabine has demonstrated activity in breast, ovarian, non-small cell lung, pancreatic, and bladder cancers [63].
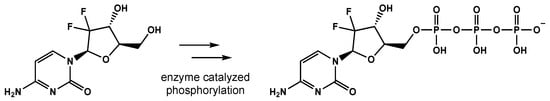
Figure 2. The conversion of gemcitabine to gemcitabine-5′-triphosphate before being incorporated into DNA and RNA, eventually leading to strand termination.
In vitro studies have determined the gemcitabine administration schedule is essential for maximal radiosensitzation. Gemcitabine achieved radiosensitization with long exposure (24 h) to low gemcitabine concentrations or brief treatments with increased concentrations [64]. Maraveyas et al conducted a phase I study in brain metastases patients evaluating the maximum tolerated dose of concomitant gemcitabine and RT [65]. A phase I study then evaluated gemcitabine with concomitant RT in newly diagnosed GBM patients [66]. In this study, gemcitabine was delivered at 10 mg/m2/min on a weekly basis for 6 weeks 24 to 72 h prior to concomitant RT (60 Gy in 30 fractions) with the identification of dose-limiting toxicity and maximum tolerated dose as the primary end-points. Based on this study, 175 mg/m2/weekly was recommended for further evaluation in a phase II study. Twenty-three patients were enrolled in their phase II study and found concomitant RT and gemcitabine were well-tolerated with few severe adverse events [67]. Additionally, disease control was observed in both methylated and unmethylated MGMT promoter tumors (91% and 77.5%, respectively).
To date, there is evidence gemcitabine has the ability to cross the blood–brain barrier [68], but some drawbacks include its short plasma half-life, adverse effects related to high drug doses (e.g., myelosuppression, thrombocytopenia, edema), and resistance related to altered expression of nucleoside transporters, kinases, and enzymes [56]. Researchers are currently exploring various delivery strategies for overcoming these limitations (e.g., encapsulation, conjugation, and convention-enhanced delivery) [69,70,71]. For example, Guo et al. surmised gemcitabine coupled to a peripheral benzodiazepine receptor ligand may enhance brain tumor uptake [70]. In their xenograft model, the conjugated agent resulted in a two-fold enhancement in brain tumor selectively compared with gemcitabine alone.
3. Kinase Inhibitors
3.1. Tyrosine Kinase Inhibitors
Tyrosine kinase inhibitors (TKIs) block receptor signaling, inhibiting cell growth and proliferation. Since the approval of imatinib in 2001 for the treatment of chronic myeloid leukemia, there has been an explosion of TKI utilization in multiple types of cancer [72]. TKIs have incredible potential for treating GBM considering their ability to block cell signaling pathways such as EGFR, PDGFR, and VEGF/VEGFR.
EGFR amplification is seen in approximately 40% of GBM cases, correlating with decreased apoptosis, increased cellular proliferation, tumorigenesis, and radioresistance [73,74,75]. Erlotinib is a TKI that has demonstrated activity against the EGFRvIII mutant receptor in preclinical models [76]. Erlotinib is a quinazoline derivative that reversibly inhibits autophosphorylation of EGFR (Figure 3) [77]. Various phase II studies have evaluated the efficacy of erlotinib with concurrent RT and TMZ, but a range of survival and toxicity outcomes have been reported. The first trial included 97 GBM patients who were given erlotinib alone for 1 week followed by concurrent erlotinib, TMZ (75 mg/m2 daily), and RT (60 Gy total) [78]. Patients had a median survival time of 15.3 months, but there was no significant benefit compared to RT/TMZ arm of the European Organization for Research and Treatment of Cancer/National Cancer Institute of Canada trial 26981/22981. Furthermore, molecular subset analysis did not reveal that EGFR amplification was predictive of survival. Another phase II trial included 27 newly diagnosed GBM patients [79]. In this trial, erlotinib was determined to be not efficacious with unacceptable toxicity (grade 3 and 4 toxicities including thrombocytopenia, anemia, lymphopenia, fatigue, and febrile neutropenia). Numerous clinical trials have evaluated other EGFR TKIs (e.g., gefitinib, afatinib) in GBM patients [79,80,81]. Unfortunately, all EGFR TKIs to date have failed to show efficacy in GBM. Researchers hypothesize the lack of efficacy may be due to poor blood–brain barrier penetration, altered signaling pathways, and/or genetic heterogeneity [82].
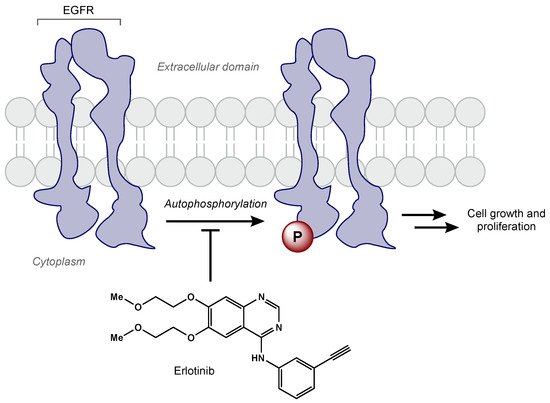
Figure 3. Erlotinib reversibly inhibits EGFR tyrosine kinase activity, which prevents cell growth and proliferation of cancer cells.
Recently, preclinical studies have tested osimertinib, a third-generation EGFR TKI, in various GBM cell lines and mice [83]. Liu et al. showed osimertinib inhibited GBM cell growth ten-fold higher than first-generation EGFR inhibitors and prolonged survival in GBM-bearing mice.
3.2. mTOR Inhibitors
Rapamycin (mTOR) is a protein kinase that is an important regulator of cell survival and proliferation [84]. mTOR is localized in two distinct multi-protein complexes called mTORC1 and mTORC2 [85]. Previous research efforts have uncovered the critical role of mTOR in GBM pathogenesis [86,87]. Recent studies have shown GSCs can activate the mTOR pathway in microglia, creating an immunosuppressive microenvironment that promotes GBM proliferation [88].
Temsirolimus was the first mTORC1 inhibitor investigated in clinical trials (Figure 4). Temsirolimus has been shown to target GICs in preclinical studies, but has failed to demonstrate clinical benefit [89]. Sirolimus, another mTOR inhibitor, also had promising preclinical results, but failed to improve survival, despite being well tolerated [90]. Everolimus, another rapamycin derivative, is a downstream regulator of the EGFR/phosphatidylinositol-3 kinase (PI3K) pathway that has demonstrated radiosensitization in preclinical studies [91]. The North Central Cancer Treatment Group (NCCTG) conducted a phase II trial where weekly everolimus was given concurrently with RT plus TMZ. Ma et al. reported moderate toxicity and survival rates similar to historical phase II trials [92]. The RTOG 0913 trial randomized 171 GBM patients to receive RT with concurrent and adjuvant TMZ with or without daily everolimus (10 mg) [93]. Chinnaiyan and colleagues reported no significant difference in PFS and inferior OS for the patients that received everolimus. There was a significant increase in treatment-related toxicity in patients that received everolimus compared with the control arm; in the experimental arm, there were greater grade 4 and 5 events (30.6% and 11.8%, respectively) than in the control arm (17.9% and 1.3%, respectively).
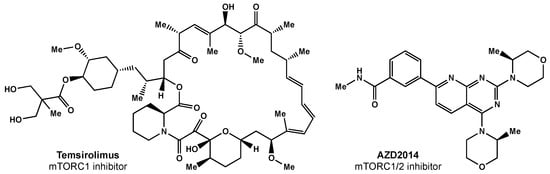
Figure 4. Small molecule inhibitors of mTOR.
Researchers surmise that the lack of efficacy may be related to everolimus only selectively inhibiting mTORC1 alone; studies have shown this inhibition can result in increased AKT activation via the activation of mTORC2 [94]. There are ongoing efforts focused on designing a suitable mTORC1/2 inhibitor [95]. AZD2014 is an inhibitor of mTORC1 and mTORC2 (Figure 4) that has shown radiosensitivity in preclinical studies [95] and is being evaluated in a phase I trial (NCT02619864).
4. Oxygen Mimetics
Conventional RT induces DNA damage via the formation of free radicals generated from the radiolysis of water. Reductants such as glutathione are able to neutralize the radical-induced damage within the cells, but if oxygen is present, this process is prevented, and the damage becomes irreversible. Hypoxic areas of solid tumors greatly hamper the effects of RT, leading researchers to seek oxygen mimetics [96].
Small molecules have been utilized as oxygen mimetics for decades [97] and have historically contained nitro groups that act as electron acceptors [98]. One of the earlier compounds that demonstrated radiosensitizing effects is misonidazole. Although imidazole showed radiosensitizing effects in murine tumors, its lipophilic properties prevented successful translation into clinical trials [99]. Derivatives of misonidazole were tested, and etanidazole had superior hydrophilicity due to the addition of an amide and hydroxyl group [100]. RRx-001 is a dinitro compound originally used as an ingredient in rocket fuel that has demonstrated radiosensitization properties with low toxicity [101]. Currently, RRx-001 is being evaluated in a phase I trial for patients with newly diagnosed glioblastoma (NCT02871843).
Hydrogen peroxide has been explored as a route for enhancing the efficacy of RT [102] and has been evaluated in a phase I/II trial (NCT02757651). Several studies also explored nicotinamide in combination with carbogen breathing in accelerated RT (ARCON) for various tumor types, including laryngeal, bladder, and head and neck [103,104,105,106]. Nicotinamide is a vasoactive agent that decreases perfusion-limited hypoxia, and carbogen (98% oxygen and 2% CO2) decreases diffusion-limited hypoxia [107]. Transfusion with red blood cells, in theory, should increase the oxygen supply of tumor cells, but this has failed to demonstrate benefit [108].
5. Reductive Agents
Bioreductive agents such as quinones and transition metal complexes have garnered attention due to their synergistic effects with RT and their preferential cytotoxicity towards hypoxic cells. Tirapazamine is a pro-drug that can be reduced to a free radical, leading to single- and double-strand DNA breaks under hypoxic environments (Figure 5) [109]. Del Rowe et al conducted a phase II study with RT plus tirapazamine [110]. Although toxicity was acceptable, tirapazamine demonstrated no survival benefit.
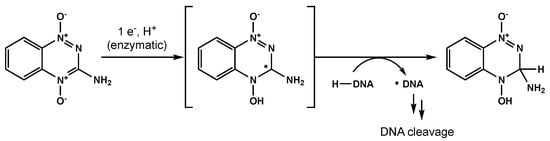
Figure 5. One proposed mechanism for tirapazamine-mediated DNA cleavage under hypoxic conditions.
An analogue of tirapazamine is SN30000 with more favorable diffusion properties and is currently under development [111,112]. Other analogues such as nimorazole demonstrated efficacy in several trials and are currently used in the treatment of head and neck cancers in Denmark [113].
6. Histone Deactylase Inhibitors
Histone deacetylases (HDACs) are enzymes that regulate chromatin structure and gene expression via deacetylation of histones and other cytoplasmic and nuclear proteins [114]. Valproic acid, an HDAC inhibitor, has demonstrated increased RT sensitivity in vitro and in vivo. Although the mechanism is unclear, researchers have proposed radiosensitization may be due to the inhibition of chromatin remodeling [115]. Krauze and colleagues conducted a phase II study evaluating the addition of valproic acid to RT plus TMZ [116]. Median OS was 29.6 months (range, 21–63.8 months), PFS was 10.5 months (range, 6.8–51.2 months), and the addition of valproic acid was generally well tolerated. The utilization of valproic acid remains controversial, though, after a pooled analysis found valproic acid at antiepilepsy doses was not associated with improved PFS or OS [117]. Vorinostat is another HDAC inhibitor that has been explored in one phase I/II trial, but failed to meet its primary efficacy end point [118].
7. Targeting DNA Repair Pathways
Ataxia-telangiectasia-mutated (ATM) serine/threonine protein kinase plays a role in the repair of DNA double-strand breaks [119]. ATM activation is induced within minutes of irradiation, and GSCs are particularly resistant following increased activation of ATM [120,121]. Carruthers et al. demonstrated GSCs display a robust intrinsic phosphor-ATM signal that is further enhanced following irradiation [121]. Other studies have found GBM cell lines and GSCs are radiosensitized by ATM inhibition [122].
Recently, medicinal chemists have developed a novel series of ATM inhibitors that demonstrate excellent efficacy and good pharmacokinetic properties [123]. AZD0156 was selected as a suitable candidate for clinical trials (NCT02588105). Further structure–activity relationship lead optimization led to the development of AZD1390, an orally bioavailable inhibitor with greater blood–brain barrier penetrance (Figure 6) [119]. A phase I clinical trial (NCT03423628) is currently recruiting GBM patients for the evaluation of AZD1390 in combination with RT.
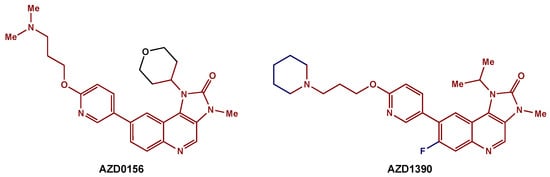
Figure 6. ATM inhibitors: AZD0156 was modified to AZD1390, an orally available compound with greater blood–brain barrier penetrance. The preserved core is highlighted in red.
8. Allosteric Modifiers of Hemoglobin
Phenoxyacetic acid compounds were initially utilized as lipid-lowering drugs but later were found to stabilize the T state of hemoglobin [124]. In a phase III trial, efaproxiral, a phenoxyacetic acid analogue, was found to enhance the effect of RT in patients with advanced lung cancer [125]. Kleinberg et al. then surmised GBM patients may benefit from the radio-enhancing effects of efaproxiral because GBM tumors are known to be hypoxic [126] and radioresistant [127]. Although the results were promising, a large dose was needed to reach a therapeutic effect, and long-term dose-related side effects are a concern [128].
9. Immunotherapy
9.1. Anti-Angiogenic Therapy
VEGF inhibitors such as bevacizumab have been explored with the hope of targeting angiogenesis [129]. Chinot et al conducted a phase III trial evaluating the addition of bevacizumab to RT (2 Gy per fraction, total of 60 Gy) plus TMZ (75 mg/m2/day for 6 weeks) in patients with newly diagnosed GBM [130]. Although there was increased PFS in the bevacizumab group vs. placebo (10.6 months vs. 6.2 months), there was not a significant difference in OS. Furthermore, there were higher rates of adverse events with bevacizumab than with the placebo. Gilbert et al. also conducted a phase III randomized trial investigating the addition of bevacizumab to RT and TMZ [131]. Their study also demonstrated improved PFS (10.7 months vs. 7.3), although the difference was not significant according to the pre-specified alpha level (p < 0.004). The authors also noted a slight increase in adverse events and, over time, a decreased quality of life and neurocognitive function in the bevacizumab group.
9.2. Immune Checkpoint Inhibitors
Cancer immunotherapy is based on the concept of immunosurveillance where the immune system can actively detect and eliminate cancer cells, but some tumor cells are able to develop the ability to evade the immune system through immunoediting [132]. Immunoediting is a process where the immune system can both constrain and promote tumor progression [133]. Researchers propose this complex dynamic occurs in three phases: Elimination (the immune system can recognize and kill transformed cells), equilibrium (tumor growth is limited), and escape (edited tumors can grow, unrestrained) [134].
Immunotherapy aims to overcome this immunoresistance with immune checkpoint inhibitors (ICIs) [135]. Immune checkpoints are crucial for self-tolerance, and cancer cells exploit this feature via the upregulation of various pathways (e.g., PD-1/PD-L1, CTLA-4) [136]. Over the past decade, ICIs have revolutionized the treatment of solid tumors and have created renewed excitement within the field of cancer immunotherapy [137].
Although radiation is known to create DNA damage, several studies have suggested the immune system may impact the efficacy of radiation [138]. The exact mechanisms dictating how radiation and the immune system interact are still unclear, but data have revealed CD8 T cells play a key role [139,140]. In theory, combining RT and checkpoint blockage immunotherapy should increase radiosensitization.
Immune checkpoint inhibitors were believed to affect the tumor microenvironment by enhancing the expression of cytokine and chemokine release, which increases immune cell infiltration [141,142]. Anti-PD-1 monoclonal antibodies have had success in the setting of hepatocellular carcinoma, non-small cell lung cancer, renal cell carcinoma, melanoma, and a variety of other solid tumors [136]. Anti-CTLA-4 monoclonal antibodies have also demonstrated a survival benefit for metastatic melanoma [143].
Unfortunately, the addition of ICIs to GBM treatment has led to disappointing initial results.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10071763
This entry is offline, you can click here to edit this entry!