Bacterial pathogens as causative agents of infection constitute an alarming concern in the public health sector. In particular, bacteria with resistance to multiple antimicrobial agents can confound chemotherapeutic efficacy towards infectious diseases. Multidrug-resistant bacteria harbor various molecular and cellular mechanisms for antimicrobial resistance. These antimicrobial resistance mechanisms include active antimicrobial efflux, reduced drug entry into cells of pathogens, enzymatic metabolism of antimicrobial agents to inactive products, biofilm formation, altered drug targets, and protection of antimicrobial targets. These microbial systems represent suitable focuses for investigation to establish the means for their circumvention and to reestablish therapeutic effectiveness.
- bacteria
- antimicrobial resistance
- infection
- pathogenesis
- multidrug resistance
1. Introduction
Bacteria as microbial pathogens are causative agents of life-threatening infectious diseases [1]. Such pathogenic bacteria produce alarming numbers in terms of morbidity and mortality outcomes [2][3]. One crucial avenue towards bacterial pathogenesis involves the reduction in the therapeutic effects of antibacterial chemotherapy [4][5]. Throughout their evolutionary history, bacterial pathogens have developed various means of resisting the inhibitory and bactericidal consequences of antimicrobial agents [4]. Such antimicrobial resistance systems involve the engagement of bacterial molecular and cellular-based machinery [6]. Interestingly, the selection of a bacterial variant with resistance to a single antimicrobial agent frequently manifests the emergence of a multidrug resistance characteristic in the new mutant [7]. Newly emerged bacterial pathogens with resistance to multiple antibacterial agents can result in compromised efficacy in the treatment of infection [3][8].
Mechanisms of antimicrobial resistance include the active export systems within the membranes of bacteria, prevention of antimicrobial entrance into cells of pathogenic bacteria, enzymatic destruction of antimicrobial agents, production of thick biofilms, modified targets of antimicrobials, and bacterial sites of action that are protected from antimicrobials, (Figure 1) [2][4]. Furthermore, multidrug-resistant bacteria have developed mechanisms that confer the DNA transfer of genetic determinants of resistance to pathogenic species in the clinical setting, the food production industry, the human gut, and in agriculture [9].
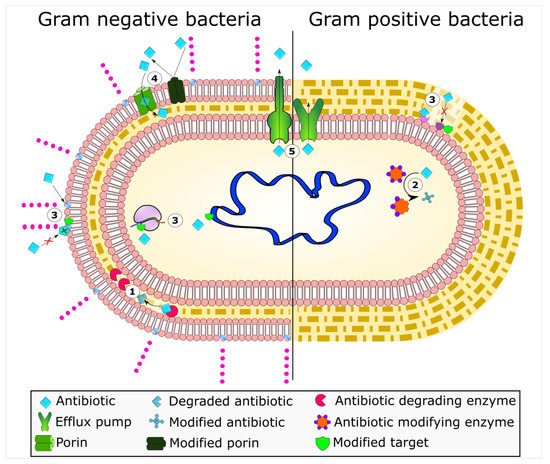
Figure 1. Bacterial mechanisms of resistance to antimicrobial agents. The common mechanisms of antibiotic resistance in bacteria are enzymatic hydrolysis (1), enzymatic modifications of antibiotics by group transfer and redox process (2), modifications of antibiotic targets (3), reduced permeability to antibiotics by modifications of porins (4), and active extrusion of antibiotics by membrane efflux pumps (5).
Thus, new strategies for the circumvention of bacterial resistance to antimicrobial agents are desired [10]. In order to discover novel approaches to address multiple antimicrobial resistances in these microbial pathogens, however, it is necessary to attain a clear understanding of these resistance systems at the molecular and cellular levels. For young and new investigators, here, we consider an introductory overview of each of these disparate bacterial resistance mechanisms here.
2. Enzyme-Based Antimicrobial-Inactivation Systems
Along the timeline of antibiotic discovery and introduction, several enzymatic mechanisms of antibiotic inactivation were also discovered. Although very few novel mechanisms of antibiotic resistance have been reported in recent times, several new variants of known enzymes that endow bacteria with resistance to newly introduced drugs have emerged, suggesting that the bacterial response to new antibiotics or the modified versions of existing antibiotics is swift. The enzymatic mechanisms of antibiotic resistance include hydrolysis, group transfer, and redox processes [4]. In terms of diversity, evolution, and spread, antibiotic resistance enzymes contribute remarkably to the bacterial ability to overcome antibiotic pressure. The β-lactamases are the oldest known and the most diverse antibiotic degrading enzymes that cleave the β-lactam ring of the penicillin group of antibiotics and render them ineffective. The first such β-lactamase was discovered soon after the first antibiotic penicillin was in clinical use. Scientific evidence suggests the existence of β-lactamases before penicillin was clinically employed, emphasizing that the production of antimicrobial compounds and the mechanisms to endure them occur in parallel in the environment [11]. Bacteria that produce antibiotics apparently require mechanisms to overcome the lethal effects of the compounds, and these are in the form of concurrent production of degradative enzymes, mutations in targets of antibiotics, or active extrusion of antibiotics from the cell so that the antibiotic-producing cell is protected. However, the selection pressure created due to the extensive use of antibiotics in humans and animals propagated the resistant clones of bacteria in clinical and food production environments. In due course of time, genetic exchange mechanisms facilitated the wider dissemination of resistance traits in bacterial communities. The introduction of more antibiotics, newer as well modified, augmented the process of evolution and spread of resistance mechanisms. Since the majority of the antibiotics introduced in the last two decades are mostly the modified versions of existing antibiotics belonging to the same classes (e.g., β-lactams), a few mutations in the enzymes could render bacteria quickly resistant to them [11].
The β-lactams constitute the largest group of clinically used antibiotics, comprising of penicillins, cephalosporins of different generations, monobactams, and carbapenems, all of which are characterized by the presence of 3-carbon, 1-nitrogen containing β-lactam ring. The β-lactam antibiotics inhibit the bacterial proteins known as penicillin-binding proteins (PBPs), which perform the critical role of peptide cross-linking during peptidoglycan cell wall biosynthesis. The structural mimicry of the d-Ala-d-Ala terminal fragment of cross-linking peptide by β-lactams facilitates competitive inhibition of PBPs [12], which stops the cell wall synthesis leading to bacterial cell lysis and death [13]. However, bacteria gain resistance to lactam antibiotics by modifying their PBPs, which are no longer susceptible to binding by the antibiotic. Alternatively, bacteria produce powerful lactamases that degrade antibiotics before they can bind with the PBPs. Since their discovery in the early 1940s, the family of β-lactamases has grown seamlessly, with more than 300 enzymes identified globally [14][15].
The early β-lactamases were penicillinase enzymes that degraded penicillin, which started appearing rapidly in clinical bacteria [16][17]. The introduction of modified, semisynthetic penicillins such as methicillin, ampicillin, and amoxicillin resulted in the gradual appearance of β-lactamases capable of degrading them. The first plasmid-borne transferrable β-lactamase was TEM-1, followed by TEM-2 and SHV-1 enzymes [18][19]. TEM is the most common mechanism of ampicillin resistance compared to less prevalent SHV-1, although both have the same affinity for this antibiotic. TEM and SHV share 60% amino acid similarity between them and are inhibited by clavulanic acid, tazobactam, and sulbactam. The discovery of cephalosporin C in the early 1960s heralded an era of synthetic cephalosporins, which was thought to fend off β-lactamases. Structurally, cephalosporins have their β-lactam ring fused to a six-membered dihydrothiazine ring compared to penicillins in which the β-lactam is fused with a five-membered thiazolidine ring [20]. Subsequently, carbapenem and monobactam groups of β-lactam antibiotics with structurally variant lactam rings were discovered from natural sources and formed the basis for the synthesis of similar compounds with modifications. However, the enzymes extended-spectrum β-lactamases (ESBLs) that could hydrolyze a wide range of cephalosporins emerged from TEM and SHV lactamases by point mutations [18]. ESBLs hydrolyze a broad spectrum of cephalosporins, including first, second, third-generation cephalosporins and aztreonam, but not cephamycins and carbapenems, and are inhibited by clavulanic acid [18][21]. As a consequence of mutations and the expansion of the substrate range, ESBLs have a lesser affinity for classical β-lactams compared to their ancestral β-lactamases. Subsequently, CTX-M type ESBLs with high affinity for cefotaxime emerged independent of TEM and SHV lactamases, and these supposedly evolved from β-lactamases of Kluyvera spp. [22]. Over the years, CTX-M has overtaken other ESBLs in terms of number and global distribution, with more than 230 types identified to date. Figure 2 shows the timeline of the evolution of β-lactamases in relation to the introduction of β-lactam antibiotics for clinical use.
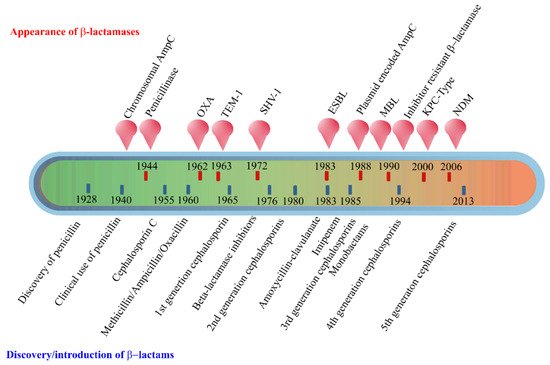
Figure 2. Evolution of β-lactamases. Within five decades of discovering the first penicillin-degrading enzyme, β-lactamases capable of hydrolyzing most β-lactam antibiotics, and resistance to inhibitors have emerged. The ability to tolerate a broad spectrum of β-lactams and inhibitor combinations is bolstered by the presence of multiple β-lactamase-encoding genes in a single pathogen.
The initial efforts to classify β-lactamases were based on their functional characteristics such as the substrate-inhibitor profiles, protein molecular weight, isoelectric point, etc. [12][14][23]. A second approach employed amino acid sequence similarities and enzymatic activities to classify β-lactamases into four main groups, of which groups A, C, and D are serine β-lactamases, while class B is composed of metallo β-lactamases that require active site zinc ion(s) for their hydrolytic activities [12][24]. Group A enzymes form the largest group of lactamases comprising some of the critical resistance enzymes such as TEM, SHV, and CTX-M type of β-lactamases. Other important ESBLs include the carbapenem hydrolyzing KPC type ESBLs originally reported from Klebsiella pneumoniae, which have an expanded substrate spectrum encompassing the cephalosporins and carbapenems but susceptible to inhibition by clavulanates and boronic acid [23][25]. The chromosomally encoded AmpC (class C) cephalosporinases described early in the timeline of the discovery of β-lactamases have no homology with penicillinases and thus constitute a distinct group of enzymes [26][27]. Commonly found in Enterobacteriaceae, AmpC enzymes are inducible and are produced at low basal levels, and preferentially hydrolyze cephalosporins including cefoxitin but not cefepime. These are generally resistant to inhibition by clavulanic acid, sulbactam, or tazobactam. The metallo-β-lactamases or MBLs belonging to class B have vigorous hydrolytic activities against carbapenems and are also active against a range of cephalosporins [28][29]. In 2009, a new variant New Delhi Metallo-β-lactamase (NDM), emerged, and since then, it has been reported from all over the world [29]. NDM confers resistance to all β-lactam antibiotics except aztreonam, and the plasmid carrying blaNDM gene harbors resistance markers for several other antibiotics. VIM and IMP are other important class B carbapenemases commonly encountered in Enterobacteriaceae.
The OXA type enzymes belonging to the Class D lactamase group were originally discovered as plasmid-encoded oxacillin hydrolyzing enzymes in lactose non-fermenting bacteria such as Pseudomonas, Acinetobacter, and Shewanella, and later in Enterobacteriaceae through plasmid exchange [30][31]. These enzymes are poorly inhibited by lactamase inhibitors such as clavulanic acid. Although OXA lactamases have a narrow substrate range composed of penicillins, cloxacillin, and oxacillin, the enzymes evolved to hydrolyze extended-spectrum cephalosporins and carbapenems through point mutations, and these abilities vary among different OXA types [28][32].
The β-lactamase mediated antimicrobial resistance is widespread among ESKAPE (Enterococcus, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and E. coli) group of organisms, infections with which are usually associated with a significantly higher economic burden and highest risk of mortalities [33][34]. The World Health Organization (WHO) has recognized carbapenem-resistant Enterobacteriaceae (CRE) as a serious global health scourge for which the development of new antimicrobials is critically needed [35].
Enzymatic hydrolysis is also a common mechanism of resistance against macrolides, rifampicin, and fosfomycin. Many Enterobacteriaceae members produce plasmid-encoded esterases EreA and EreB that hydrolyze the macrolactone ring of 14- and 15-membered macrolides such as erythromycin A, clarithromycin, and azithromycin [36][37]. The structurally altered macrolide antibiotic will no longer be able to bind to its preferred target site in the ribosome [38].
Another important mechanism of enzymatic degradation is associated with the manganese ion (Mn2+)-dependent, chromosomally-encoded FosX that uses water to cleave the epoxide ring of fosfomycin. Other fosfomycin modifying metalloenzymes include FosA, FosB, and two epoxide kinases FomA and FomB [39]. FosA is a Mn2+ and K+-dependent glutathione-S-transferase, while FosB is a Mg2+ thiol-S-transferase. The mechanism involves adding glutathione or thiol groups to the oxirane ring of fosfomycin resulting in an inactive drug [40]. FomA and FomB kinases utilize ATP and Mn2+ ions to phosphorylate the oxirane ring of fosfomycin [39].
Tetracyclines are in use for over 70 years as widely used antibiotics in human and animal medicine [41]. Tetracycline is broken down by a monooxygenase enzyme Tet(X), which is oxygen- and FAD-dependent [42]. Tet(X) monohydroxylates break down tetracyclines at position 11a, followed by non-enzymatic degradation. Similarly, enzymatic monoxygenation of the naphthyl group of rifamycin antibiotics by monooxygenases (Rox) inactivate them by leading to the linearization of the naphthoquinone or naphthohydroquinone ring [43].
Enzymatic modification of antibiotics by the transfer of functional groups, such as acyl, glycosyl, ribosyl, nucleotidyl, phosphoryl, or thiol groups, confers resistance to a range of antibiotics, including aminoglycosides, rifamycins, macrolides, epoxides, and chloramphenicol [44]. The aminoglycoside modifying enzymes (AME) responsible for resistance to different aminoglycoside antibiotics include N-acetyltransferases (AAC), O-adenyltransferases (ANT), and O-phosphotransferases (APH). These enzymes catalyze the modification of various hydroxyl or the amino groups of the aminoglycosides resulting in their inability to bind to their 30S ribosomal targets [45]. Similarly, in Gram-negative bacteria, a plasmid-encoded ADP-ribosyltransferase (Arr-2) is commonly responsible for rifampin resistance [46]. Similarly, chloramphenicol is modified by acetyl-CoA-dependent acetylation of its 3-hydroxyl group by chloramphenicol acetyltransferase (CAT) enzymes [47]. The modified antibiotic does not bind to its target site, the 50S subunit of ribosomes. CATs are widely distributed among Gram-positive and -negative bacteria and show little amino acid sequence similarities, with only 25 amino acid residues conserved among all CAT variants [47].
3. Alteration of Antimicrobial Targets
As bacterial enzymes mentioned above alter drug structures, the drug targets may likewise be altered, preventing drug binding and, thus, conferring resistance. Antimicrobial targets play vital roles in microbial growth or survival and, thus, serve as potentially useful targets for mitigating infection. In addition, these targets must differ or be completely absent from humans or the animal species being treated with an antimicrobial to allow for a selective mode of action. A classic example of such a target is peptidoglycan. Peptidoglycan is essential to the growth and survival of many bacterial species and has a chemical structure that is not present in the mammalian hosts they infect. This allows for the targeting of enzymes responsible for the synthesis and assembly of peptidoglycan. The function of proteins associated with these target sites makes it non-viable for a bacterium to evolve resistance by removing these proteins. However, mutations that allow for continued functionality while reducing the ability of an antimicrobial agent to bind them at the target site have been a veritable regularity in the arms race between antimicrobial substances and antimicrobial-resistant bacteria. In addition to peptidoglycan, alteration in target sites has been attributed to ribosomes, nucleic acid enzymes, and lipopolysaccharides [48].
As discussed previously in this review, peptidoglycan inhibition by glycopeptides involves the binding of the peptidyl-d-alanyl-d-alanine terminus of peptidoglycan precursors. This binding prevents integration via the transglycosylase activity of these precursors into the cell wall [49], as shown in Figure 3.
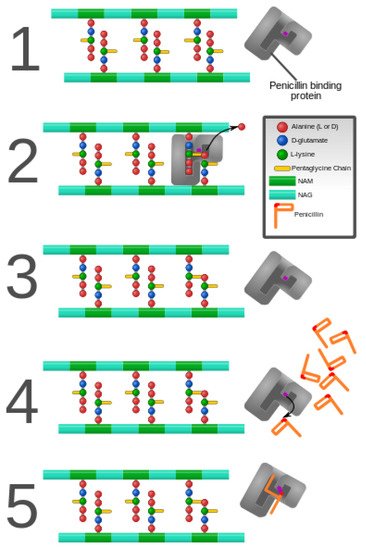
Figure 3. Penicillin and penicillin-binding protein of the bacterial cell wall. (1) The peptidoglycan layer of a bacterial cell wall harbors the repeating moieties of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM). The NAM subunits bind short variable peptide chains, usually l-Ala and two distal d-Ala residues. (2) The PBP cross-links the peptide side chain, releasing a free Ala. (3) Upon cross-linking, the PBP dissociates from the cell wall. (4) Penicillin binds the PBP active site, affecting its enzyme activity. (5) The β-lactam ring of penicillin is cleaved during its reaction with PBP. Penicillin stays covalently bound PBP, permanently inhibiting the active site. Altered PBPs, such as PBP2a, are unable to accommodate penicillin-binding, preventing cell wall synthesis inhibition [48][49].
PCBs are one mechanism for antimicrobial resistance, but the peptidoglycan precursors themselves can undergo alteration, which reduces the affinity of antimicrobials without the involvement of enzymatic inactivation. Such is the case with Enterococcus faecium and E. faecalis, which have been discussed in the literature as developing resistance by acquiring one of two related gene clusters encoding VanA and VanB [50][51]. These gene clusters produce a modified terminus that contains d-alanyl-d-lactate as opposed to d-alanyl-d-alanine [50]. This alteration leads to glycopeptides having a much lower binding affinity [52]. Thus, these gene clusters, found on transposable elements, have allowed the spread of modified targets in enterococci. Similarly, there are rarer but related gene clusters that have been shown to modify peptidoglycan precursors, such as those encoding VanD [53], VanE [54], and Van G [55].
Ribosomes, serving the vital role of protein synthesis, are common to both prokaryotic and eukaryotic organisms but differ quite vastly from one another in structure, making them another suitable candidate for antimicrobial targeting [56]. The 50S ribosomal unit serves as the binding site for macrolide, lincosamide, and streptogramin B [57]. Recalcitrance to these specific antimicrobials is known as MLS(B) type resistance [57], and it results from a post-transcriptional modification of the 23S rRNA component of the 50S ribosomal subunit that is involved with methylation or dimethylation of key adenine bases in the peptidyl transferase functional domain [58]. Mutations in the 23S rRNA, close to the site of methylation have also been associated with resistance to the macrolide group of antibiotics in a range of organisms, such as Helicobacter pylori [59] and propionibacteria [60]. Macrolide resistance in S. pneumoniae has been attributed to an alteration in the L4 and L22 proteins of the 50S subunit [61][62]. Oxazolidinones bind to the 50S subunit but have a more complex set of interactions associated with their mechanism of action [63]. The translocation of peptidyl-tRNA from the A site to the P site is hindered by this class of antibiotics, but enterococci have been documented to have an altered the P site through the substitution of U in place of G in the peptidyl transferase region (position 2576) of the 23S rRNA, thus resulting in a lowered binding affinity in the 50S subunit for this class of antibiotics [64][65][66]. Mutations more closely associated with the A site have been found in E. coli at positions 2032 and 2447 which confer resistance to the oxazolidinone drug linezolid [67].
The 30S ribosomal unit is the target of tetracycline and of aminoglycosides, which function by preventing the decoding of mRNA [68]. Mutations of the gene encoding 16S rRNA confer resistance to this class of antimicrobials [69]. Suzuki and colleagues discovered that substitutions at positions 1400, 1401, and 1483 led to kanamycin resistance in clinical isolates of Mycobacterium, and further strengthened the claim that these changes led to resistance by identifying their absence in kanamycin-sensitive Mycobacterium isolates [70]. Position 1400 was the most common substitution, featuring an A to G change [70]. The same A to G substitution at position 1408 led to high resistance against amikacin, kanamycin, gentamicin, tobramycin, and neomycin in clinical isolates of Mycobacterium abscessus [71].
Active Efflux Pumps of Antimicrobial Agents
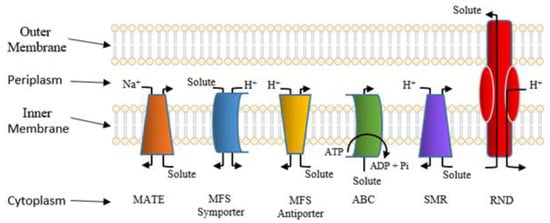
Future Directions
8. Concluding Remarks
This entry is adapted from the peer-reviewed paper 10.3390/antibiotics10050593
References
- Ziebuhr, W.; Ohlsen, K.; Karch, H.; Korhonen, T.; Hacker, J. Evolution of bacterial pathogenesis. Cell. Mol. Life Sci. CMLS 1999, 56, 719–728.
- Walsh, C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775–781.
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073.
- Kumar, S.; Varela, M.F. Molecular mechanisms of bacterial resistance to antimicrobial agents. Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education. In Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2013; pp. 522–534.
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51.
- Wright, G.D. Molecular mechanisms of antibiotic resistance. Chem. Commun. 2011, 47, 4055–4061.
- Hughes, D. Selection and Evolution of Resistance to Antimicrobial Drugs. Iubmb Life 2014, 66, 521–529.
- Vivas, R.; Barbosa, A.A.T.; Dolabela, S.S.; Jain, S. Multidrug-Resistant Bacteria and Alternative Methods to Control Them: An Overview. Microb. Drug Resist. 2019, 25, 890–908.
- Silbergeld, E.K.; Graham, J.; Price, L.B. Industrial Food Animal Production, Antimicrobial Resistance, and Human Health. Annu. Rev. Public Health 2008, 29, 151–169.
- Varela, M.F.; Kumar, S. Strategies for Discovery of New Molecular Targets for Anti-Infective Drugs. Curr. Opin. Pharm. 2019, 48, 57–68.
- Pollock, M.R. Origin and Function of Penicillinase: A Problem in Biochemical Evolution. Br. Med. J. 1967, 4, 71–77.
- Bush, K.; Jacoby, G.A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976.
- Abraham, E.P. A Retrospective View of β-Lactamases. J. Chemother. 1991, 3, 67–74.
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62.
- Naas, T.; Oxacelay, C.; Nordmann, P. Identification of CTX-M-Type Extended-Spectrum-β-Lactamase Genes Using Real-Time PCR and Pyrosequencing. Antimicrob. Agents Chemother. 2007, 51, 223–230.
- Abraham, E.P.; Chain, E. An Enzyme from Bacteria Able to Destroy Penicillin. 1940. Rev. Infect. Dis. 1988, 10, 677–678.
- Rammelkamp, C.H.; Maxon, T. Resistance of Staphylococcus aureus to the Action of Penicillin. Proc. Soc. Exp. Biol. Med. 1942, 51, 386–389.
- Paterson, D.L.; Bonomo, R.A. Extended-Spectrum β-Lactamases: A Clinical Update. Clin. Microbiol. Rev. 2005, 18, 657–686.
- Sirot, J.; Chanal, C.; Petit, A.; Sirot, D.; Labia, R.; Gerbaud, G. Klebsiella pneumoniae and Other Enterobacteriaceae Producing Novel Plasmid-Mediated β-Lactamases Markedly Active against Third-Generation Cephalosporins: Epidemiologic Studies. Rev. Infect. Dis. 1988, 10, 850–859.
- Abraham, E.P.; Newton, G.G. The Structure of Cephalesporin C. Biochem. J. 1961, 79, 377–393.
- González-Candelas, F.; Comas, I.; Martínez, J.; Luis; Galán, J.; Carlos; Baquero, F. 12—The Evolution of Antibiotic Resistance. In Genetics and Evolution of Infectious Disease; Tibayrenc, M., Ed.; Elsevier: London, UK, 2011; pp. 305–337. ISBN 978-0-12-384890-1.
- Humeniuk, C.; Arlet, G.; Gautier, V.; Grimont, P.; Labia, R.; Philippon, A. β-Lactamases of Kluyvera ascorbata, Probable Progenitors of Some Plasmid-Encoded CTX-M Types. Antimicrob. Agents Chemother. 2002, 46, 3045–3049.
- Bush, K. Bench-to-Bedside Review: The Role of β-Lactamases in Antibiotic-Resistant Gram-Negative Infections. Crit. Care 2010, 14, 224.
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A Functional Classification Scheme for β-Lactamases and Its Correlation with Molecular Structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233.
- Davidson, A.L.; Dassa, E.; Orelle, C.; Chen, J. Structure, Function, and Evolution of Bacterial ATP-Binding Cassette Systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364.
- Edlund, T.; Grundström, T.; Normark, S. Isolation and Characterization of DNA Repetitions Carrying the Chromosomal β-Lactamase Gene of Escherichia coli K-12. Mol. Gen. Genet. 1979, 173, 115–125.
- Jaurin, B.; Grundström, T. AmpC Cephalosporinase of Escherichia coli K-12 Has a Different Evolutionary Origin from That of β-Lactamases of the Penicillinase Type. Proc. Natl. Acad. Sci. USA 1981, 78, 4897–4901.
- Queenan, A.M.; Bush, K. Carbapenemases: The Versatile β-Lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458.
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a New Metallo-β-Lactamase Gene, Bla(NDM-1), and a Novel Erythromycin Esterase Gene Carried on a Unique Genetic Structure in Klebsiella pneumoniae Sequence Type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054.
- Antunes, N.T.; Fisher, J.F. Acquired Class D β-Lactamases. Antibiotics 2014, 3, 398–434.
- Evans, B.A.; Amyes, S.G.B. OXA β-Lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263.
- Afzal-Shah, M.; Woodford, N.; Livermore, D.M. Characterization of OXA-25, OXA-26, and OXA-27, Molecular Class D β-Lactamases Associated with Carbapenem Resistance in Clinical Isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2001, 45, 583–588.
- Founou, R.C.; Founou, L.L.; Essack, S.Y. Clinical and Economic Impact of Antibiotic Resistance in Developing Countries: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, 0189621.
- Zhen, X.; Lundborg, C.S.; Sun, X.; Hu, X.; Dong, H. Economic burden of antibiotic resistance in ESKAPE organisms: A systematic review. Antimicrob. Resist. Infect. Control 2019, 8, 137.
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y. Discovery, Research, and Development of New Antibiotics: The Who Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet. Infect. Dis. 2018, 18, 318–327.
- Barthelemy, P.; Autissier, D.; Gerbaud, G.; Courvalin, P. Enzymic Hydrolysis of Erythromycin by a Strain of Escherichia coli. J. Antibiot. 1984, 37, 1692–1696.
- Morar, M.; Pengelly, K.; Koteva, K.; Wright, G.D. Mechanism and Diversity of the Erythromycin Esterase Family of Enzymes. Biochemistry 2012, 51, 1740–1751.
- Golkar, T.; Zieliński, M.; Berghuis, A.M. Look and Outlook on Enzyme-Mediated Macrolide Resistance. Front. Microbiol. 2018, 9.
- Castaneda-Garcia, A.; Blazquez, J.; Rodriguez-Rojas, A. Molecular mechanisms and clinical impact of acquired and intrinsic fosfomycin resistance. Antibiotics 2013, 2, 217–236.
- Silver, L.L. Fosfomycin: Mechanism and Resistance. Cold Spring Harb. Perspect. Med. 2017, 7, a025262.
- Walsh, C. Antibiotics: Actions, Origins, Resistance; American Society for Microbiology: Washington, DC, USA, 2003.
- Yang, W.; Moore, I.F.; Koteva, K.P.; Bareich, D.C.; Hughes, D.W.; Wright, G.D. TetX Is a Flavin-Dependent Monooxygenase Conferring Resistance to Tetracycline Antibiotics. J. Biol. Chem. 2004, 279, 52346–52352.
- Koteva, K.; Cox, G.; Kelso, J.K.; Surette, M.D.; Zubyk, H.L.; Ejim, L.; Stogios, P.; Savchenko, A.; Sørensen, D.; Wright, G.D. Rox, a Rifamycin Resistance Enzyme with an Unprecedented Mechanism of Action. Cell Chem. Biol. 2018, 25, 403–412.e5.
- Wright, G.D. Bacterial Resistance to Antibiotics: Enzymatic Degradation and Modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470.
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. Medchemcomm 2016, 7, 11–27.
- Baysarowich, J.; Koteva, K.; Hughes, D.W.; Ejim, L.; Griffiths, E.; Zhang, K.; Junop, M.; Wright, G.D. Rifamycin Antibiotic Resistance by ADP-Ribosylation: Structure and Diversity of Arr. Proc. Natl. Acad. Sci. USA 2008, 105, 4886–4891.
- Shaw, W.V. Chloramphenicol Acetyltransferase: Enzymology and Molecular Biology. Crit. Rev. Biochem. 1983, 14, 1–46.
- Lambert, P.A. Bacterial Resistance to Antibiotics: Modified Target Sites. Adv. Drug Deliv. Rev. 2005, 57, 1471–1485.
- Reynolds, P.E. Structure, Biochemistry and Mechanism of Action of Glycopeptide Antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 1989, 8, 943–950.
- Bugg, T.D.; Wright, G.D.; Dutka-Malen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Molecular Basis for Vancomycin Resistance in Enterococcus faecium Bm4147: Biosynthesis of a Depsipeptide Peptidoglycan Precursor by Vancomycin Resistance Proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415.
- Billot-Klein, D.; Gutmann, L.; Sable, S.; Guittet, E.; Van Heijenoort, J. Modification of Peptidoglycan Precursors Is a Common Feature of the Low-Level Vancomycin-Resistant Vanb-Type Enterococcus D366 and of the Naturally Glycopeptide-Resistant Species Lactobacillus casei, Pediococcus pentosaceus, Leuconostoc mesenteroides, and Enterococcus gallinarum. J. Bacteriol. 1994, 176, 2398–2405.
- Cooper, M.A.; Fiorini, M.T.; Abell, C.; Williams, D.H. Binding of Vancomycin Group Antibiotics to D-Alanine and d-Lactate Presenting Self-Assembled Monolayers. Bioorganic Med. Chem. 2000, 8, 2609–2616.
- Perichon, B.; Reynolds, P.; Courvalin, P. Vand-Type Glycopeptide-Resistant Enterococcus faecium Bm4339. Antimicrob. Agents Chemother. 1997, 41, 2016–2018.
- Fines, M.; Perichon, B.; Reynolds, P.; Sahm, D.F.; Courvalin, P. Vane, a New Type of Acquired Glycopeptide Resistance in Enterococcus faecalis Bm4405. Antimicrob. Agents Chemother. 1999, 43, 2161–2164.
- McKessar, S.J.; Berry, A.M.; Bell, J.M.; Turnidge, J.D.; Paton, J.C. Genetic Characterization of VanG, a Novel Vancomycin Resistance Locus of Enterococcus faecalis. Antimicrob. Agents Chemother. 2000, 44, 3224–3228.
- Champney, W.S. Bacterial Ribosomal Subunit Assembly Is an Antibiotic Target. Curr. Top. Med. Chem. 2003, 3, 929–947.
- Weisblum, B. Erythromycin Resistance by Ribosome Modification. Antimicrob. Agents Chemother. 1995, 39, 577–585.
- Vannuffel, P.; Di Giambattista, M.; Morgan, E.A.; Cocito, C. Identification of a Single Base Change in Ribosomal RNA Leading to Erythromycin Resistance. J. Biol. Chem. 1992, 267, 8377–8382.
- Wang, G.; Taylor, D.E. Site-Specific Mutations in the 23s rRNA Gene of Helicobacter pylori Confer Two Types of Resistance to Macrolide-Lincosamide-Streptogramin B Antibiotics. Antimicrob. Agents Chemother. 1998, 42, 1952–1958.
- Ross, J.I.; Eady, E.A.; Cove, J.H.; Jones, C.E.; Ratyal, A.H.; Miller, Y.W.; Vyakrnam, S.; Cunliffe, W.J. Clinical Resistance to Erythromycin and Clindamycin in Cutaneous Propionibacteria Isolated from Acne Patients is Associated with Mutations in 23s rRNA. Antimicrob. Agents Chemother. 1997, 41, 1162–1165.
- Reinert, R.R.; Wild, A.; Appelbaum, P.; Lutticken, R.; Cil, M.Y.; Al-Lahham, A. Ribosomal Mutations Conferring Resistance to Macrolides in Streptococcus pneumoniae Clinical Strains Isolated in Germany. Antimicrob. Agents Chemother. 2003, 47, 2319–2322.
- Canu, A.; Malbruny, B.; Coquemont, M.; Davies, T.A.; Appelbaum, P.C.; Leclercq, R. Diversity of Ribosomal Mutations Conferring Resistance to Macrolides, Clindamycin, Streptogramin, and Telithromycin in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2002, 46, 125–131.
- Swaney, S.M.; Aoki, H.; Ganoza, M.C.; Shinabarger, D.L. The Oxazolidinone Linezolid Inhibits Initiation of Protein Synthesis in Bacteria. Antimicrob. Agents Chemother. 1998, 42, 3251–3255.
- Auckland, C.; Teare, L.; Cooke, F.; Kaufmann, M.E.; Warner, M.; Jones, G.; Bamford, K.; Ayles, H.; Johnson, A.P. Linezolid-Resistant Enterococci: Report of the First Isolates in the United Kingdom. J. Antimicrob. Chemother. 2002, 50, 743–746.
- Prystowsky, J.; Siddiqui, F.; Chosay, J.; Shinabarger, D.L.; Millichap, J.; Peterson, L.R.; Noskin, G.A. Resistance to Linezolid: Characterization of Mutations in rRNA and Comparison of Their Occurrences in Vancomycin-Resistant Enterococci. Antimicrob. Agents Chemother. 2001, 45, 2154–2156.
- Eliopoulos, G.M.; Meka, V.G.; Gold, H.S. Antimicrobial resistance to linezolid. Clin. Infect. Dis. 2004, 39, 1010–1015.
- Xiong, L.; Kloss, P.; Douthwaite, S.; Andersen, N.M.; Swaney, S.; Shinabarger, D.L.; Mankin, A.S. Oxazolidinone Resistance Mutations in 23s rRNA of Escherichia coli Reveal the Central Region of Domain V as the Primary Site of Drug Action. J. Bacteriol. 2000, 182, 5325–5331.
- Recht, M.I.; Douthwaite, S.; Dahlquist, K.D.; Puglisi, J.D. Effect of Mutations in the A Site of 16 s rRNA on Aminoglycoside Antibiotic-Ribosome Interaction. J. Mol. Biol. 1999, 286, 33–43.
- Recht, M.I.; Puglisi, J.D. Aminoglycoside Resistance with Homogeneous and Heterogeneous Populations of Antibiotic-Resistant Ribosomes. Antimicrob. Agents Chemother. 2001, 45, 2414–2419.
- Suzuki, Y.; Katsukawa, C.; Tamaru, A.; Abe, C.; Makino, M.; Mizuguchi, Y.; Taniguchi, H. Detection of Kanamycin-Resistant Mycobacterium tuberculosis by Identifying Mutations in the 16S rRNA Gene. J. Clin. Microbiol. 1998, 36, 1220–1225.
- Prammananan, T.; Sander, P.; Brown, B.A.; Frischkorn, K.; Onyi, G.O.; Zhang, Y.; Böttger, E.C.; Wallace, J.R., Jr. A Single 16S Ribosomal RNA Substitution Is Responsible for Resistance to Amikacin and Other 2-Deoxystreptamine Aminoglycosides in Mycobacterium abscessus and Mycobacterium chelonae. J. Infect. Dis. 1998, 177, 1573–1581.