Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Radiolabeled liposomes have attracted new interest as probes to identify the most suitable patients for treatment with liposomal formulations of common chemotherapeutics. The use of ligands for the delivery of radiotherapeutics to a specific target is still the most appealing strategy for treating tumors. The most appropriate ligand can be identified by virtually simulating its interaction with the receptor. All strategies showed great potential for use in targeted radionuclide therapy.
- targeted radionuclide therapy
- liposomes
- avidin–biotin
- docking
1. Introduction
The goal of cancer therapy is to target and destroy tumor cells without damaging healthy tissues, but chemotherapy and external beam radiotherapy (EBRT)—the main alternatives to surgical treatment—are often nonspecific or associated with toxicity [1]. Many efforts have consequently been made to find ways to deliver cancer treatments more precisely. One example is targeted radionuclide therapy (TRT), a method whereby radionuclides are carried to tumor cells by molecules with a high affinity for the target [2]. Delivering radionuclides more effectively brings many advantages. For a start, the dose of isotope administered can be modulated, reducing patients’ exposure to radiation and the costs of the treatment. Second, a high affinity for the target can reduce background activity, thereby improving imaging quality and drug tolerability. Third, the opportunity to obtain information about a drug’s biodistribution using a diagnostic agent enables the planning of patient-specific treatments. To obtain the abovementioned advantages, the tracer is administered, at first, labeled with positron or γ-emitting radionuclides (e.g., 18F, 68Ga, or 111In) to see the areas where the tracer is deposited, identify any off-target uptake [3], and predict the dose absorbed by the tumor. Then the same tracer is administered again after labeling with β-emitting radionuclides (e.g., 90Y, 177Lu, or 131I).
Alpha-emitting radionuclides and Auger electron emitters can also be used for therapy, but β-emitting radionuclides are considered ideal for treating large tumors. This is because their long-range radiation can affect neighboring cells, as well as those being targeted (crossfire effect). Alpha-emitters are short-range, high-energy emitters more suitable for treating micrometastases and blood or bone marrow malignancies. Auger electron emitters are better suited to targeting single cells [1].
The choice of radiopharmaceutical agent is of primary importance in TRT. Carrier molecules should have a high affinity and specificity for the target, they should not be toxic or immunogenic, they should be stable before and after administration, they should be capable of binding a variety of radionuclides effectively, and they should be readily available at low cost [2].
Monoclonal antibodies (MoAbs) are among the most often used targeting agents because they can recognize a specific target and be bound directly to a radionuclide. They also have several drawbacks, however, such as a large size and slow kinetics [2]. The fact that some radionuclides decay rapidly has made it necessary to reduce the circulation time of MoAbs, and this has prompted the development of engineered antibody fragments, such as single-domain antibodies, diabodies, minibodies, protein scaffolds, and more complex specific antibodies [4]. Smaller antibodies have better pharmacokinetics and a good tumor penetration. They also have dimensions below the renal filtration cutoff; hence, they can be cleared through the kidneys, which are less radioresistant than the liver (the main site of MoAb accumulation) [4].
The high expression of peptide receptors on the surface of tumors means that peptide analogs are also good targeting agents [2]. Somatostatin receptors have been used as targets for over 20 years [5], especially for the treatment of neuroendocrine tumors. The most often used somatostatin analogs are dodecanetetraacetic acid phenylalanine-1 tyrosine 3-octreotide (DOTA-TOC) and dodecanetetraacetic acid tyrosine 3-octreotate (DOTA-TATE). They are labeled with 90Y and 177Lu for treatment purposes, and with 68Ga or 111In or 99mTc for pretreatment imaging. The advantages of using peptide analogs include a well-established conjugation chemistry, an efficient penetration in solid tumors, and lower production costs [1].
Small molecules like hormones, steroids, and neurotransmitters that are internalized by specific receptors can also be used as targeting agents. An example is metaiodobenzylguanidine (MIBG), a structural analog of the neurotransmitter norepinephrine, which can be labeled with 131I or 123I for use in treating or imaging in patients with relapsing or refractory neuroblastoma, neuroendocrine tumors, or medullary thyroid cancers [1].
Despite this variety of promising targeting agents, the clinical efficiency of TRT remains low for solid tumors because the targeting agents become distributed mainly in the outer part of the tumor mass, with less radiation reaching the inner part [2].
2. Liposomes
Liposomes are nanosized vesicles consisting of a lipid bilayer that can also contain cholesterol. They have an aqueous core and can be filled with either hydrophobic drugs (encapsulated in the bilayer) or hydrophilic drugs (encapsulated in the aqueous core). By means of a lipid chain, the main molecules (e.g., drugs and targeting agents) can also be linked to the membrane surface during liposome manufacture or via post-synthesis [6]. Embedding drugs in liposomes improves their properties, achieving a better biodistribution and a lower toxicity [7], and that is why these liposomal vesicles are often used for conventional drug delivery.
Liposomes were found to accumulate at tumor sites thanks to the enhanced permeability and retention (EPR) effect. They are easily taken up by the reticuloendothelial system (RES), however [7], consequently accumulating in organs such as the liver and spleen. To avoid their rapid clearance, polyethylene glycol (PEG) chains can be conjugated to the liposome surface to extend their circulation time and enhance their accumulation at tumor sites [7].
Studies on liposomes as drug delivery agents for use with radionuclides began in the last decade and led to the synthesis of a liposomal imaging tool called Vescan, which was never commercialized as it proved unable to detect tumors [8][9].
Attention has now shifted from the radiolabeling of empty liposomes to the radiolabeling of liposomal formulations of conventional chemotherapeutics. The aims of these contents on this topic are to identify the pharmacokinetic (PK) properties of liposomal formulations once injected in vivo, and to establish which patients will better respond to this therapy.
Although liposomes are not a perfect example of a TRT, examining progress made in research on these vesicles can probably help people to better understand their potential future uses. Here are 16 articles on the radiolabeling of liposomal formulations of conventional chemotherapeutics: nine studies dealing with the pharmacokinetics of different liposomal formulations, and seven studies dealing with the patients’ different responses to the administration of liposomal formulations of chemotherapeutics.
The pharmacokinetic properties of liposomes may be influenced by the presence of a targeting agent on their surface. To give an example, Du and coworkers [10] synthesized liposomes functionalized with MoAbs against programmed cell death-1 (PD-1), a receptor selectively expressed in triple-negative breast cancer. These liposomes were then filled with doxorubicin (DOX) and dual-labeled with a fluorophore (IRDye800WC) and a radionuclide (64Cu). They proved better able to target and to treat the tumor due to the simultaneous effect of the MoAbs against PD-1 both as a targeting agent for liposomes and as an adjuvant immunotherapy for doxorubicin.
Alongside the presence of the targeting agent, the composition of a liposome may also influence its pharmacokinetic properties. Silva and coworkers [11] demonstrated that long-circulating, pH-sensitive liposomes (SpHL) containing [99mTc] DOX accumulated more in the tumor and were less active in the spleen and liver than liposomes that were not pH-sensitive. That said, Monteiro and coworkers [12] noted that the presence of folate on the surface of SpHL (filled with paclitaxel) may lead to an even more sustained and higher tumor-to-muscle ratio than in the case of nonfunctionalized liposomes.
In addition to the presence of a targeting agent, other physical characteristics may enhance liposome delivery. For instance, Yang and coworkers [7] were able to obtain a good tumor brain delivery of their liposomal formulation of DOX, with a high tumor-to-contralateral brain ratio. They associated the presence of a targeting agent (AP-1, a peptide capable of binding IL-4 receptor) with the focused ultrasound technique, which enables a temporarily disruption of the blood–brain barrier. Reversible electroporation may also enhance delivery to the tumor, with or without any targeting agent on the liposome’s surface; this technique enhances vascular permeability, altering the EPR effect and, thus, leading to a greater liposome deposition at the tumor site [13].
To better study liposome distribution, the fluorescence technique can be associated with imaging, using positron emission tomography (PET), as in the earlier-mentioned work by Du et al. [10]. Li and coworkers [6] also succeeded in developing liposomes suitable for this application; their formulation could be labeled with the fluorophore IRDye-DSPE and the radionuclides 99mTc, 186/188Re, or 64Cu thanks to the presence of DOTA on the liposome’s surface [6].
Luo and coworkers [14] demonstrated that adding porphyrin phospholipid to the liposome’s bilayer may also be useful for the development of liposomal vesicles suitable for multimodality imaging.
Double radiolabeling is another way to obtain more information about the final target of both the liposome and the encapsulated drug. The feasibility of this technique was demonstrated by Lamichhane and coworkers [15], who labeled the liposome’s surface with 111In and the carboplatin derivative it encapsulated with 18F. More attention has also been paid in recent times to the search for new radiotracers compatible with the half-life of liposomes, and 52Mn has been identified as a suitable radionuclide for this purpose [16].
Pharmacokinetic studies have revealed a marked variability in liposome uptake by different tumors. This may be linked to the tumor’s mass, as Lin and coworkers [17] found in their study; small tumors showed growth inhibition with all the treatment regimens tested (liposomes containing chemotherapeutics and/or radionuclides), whereas the growth of large tumors was only significantly inhibited by a combination of chemo- and radiotherapy. It is not unusual to see a different liposome uptake in different patients with the same tumor or different tumors in the same patient. Tumor deposition is due mainly to the EPR effect, which could complicate pretreatment planning and hamper predictions regarding a patient’s prognosis [18].
The abovementioned studies on the pharmacokinetic properties of radiolabeled liposomes enabled tumor deposition and distribution to be quantified [19], making it possible to identify patients mostly likely to respond to a liposomal therapy. Before testing liposomes in humans, it was important to demonstrate the feasibility of radiolabeling preformed liposomal formulations. This was the goal of a study by Edmonds and coworkers [18], who successfully labeled liposomal formulations of drugs containing metal-binding motifs (e.g., doxorubicin and alendronate) with PET isotopes (e.g., 89Zr, 52Mn, and 64Cu) using metal ionophores (e.g., hydroxyquinoline).
The uptake of liposomal formulations can also be studied by recreating liposomes with the same lipid composition. This was achieved in vivo by Ito and coworkers [20], who synthesized liposomes with the same lipid composition as Doxil (a liposomal formulation of doxorubicin); they found a correlation between the therapeutic effect of Doxil and a histological factor associated with the EPR effect.
Clinical studies on the biodistribution of liposomal formulations of chemotherapeutics in patients were made by Arietta and coworkers [21][22] and Lee and coworkers [23]. Arietta’s group examined the antitumor activity of a therapy combining liposomal doxorubicin (LD) with cisplatin in patients with malignant pleural mesothelioma. They labeled the LD with 99mTc and found that patients who showed a 99mTc-LD uptake of 75% or more had significantly better rates of response, progression-free survival, and overall survival than patients with uptake levels below 75%. The authors concluded that 99mTc-LD uptake could be an important biomarker for use in assessing the results of therapy with LD and cisplatin [21][22]. Lee’s group radiolabeled MM-302, an HER2-targeted Doxil formulation, with 64Cu. After promising preliminary in vitro results [19], the liposomal vesicles were administered in humans [23], and the 64Cu-MM-302 uptake was found to vary considerably, both across multiple lesions in the same patient and across different patients. A high uptake in the liver was due to the physiological metabolism of liposomes.
3. Avidin–Biotin Interaction
Avidin is a 66 kDa highly glycosylated, positively charged protein (isoelectric point~10) derived from egg white. It is tetrameric, and each monomer has a strong affinity for biotin (Kd = 10−15) [24][25][26].
The strength of the avidin–biotin interaction is such that it is considered irreversible, and this explains why its applications have been the object of so much interest. For example, it has been studied in the sphere of tumor-targeted therapy for use in a pretargeting approach, which consists of delivering MoAbs and radionuclides separately. The radionuclide delivery is delayed until the MoAbs have reached the maximum tumor-to-normal tissue ratio [27], and the avidin–biotin interaction ensures the binding of the radiolabeled agent to the previously delivered antibody [28]. Two- or three-step protocols have been used in this setting (Figure 1).
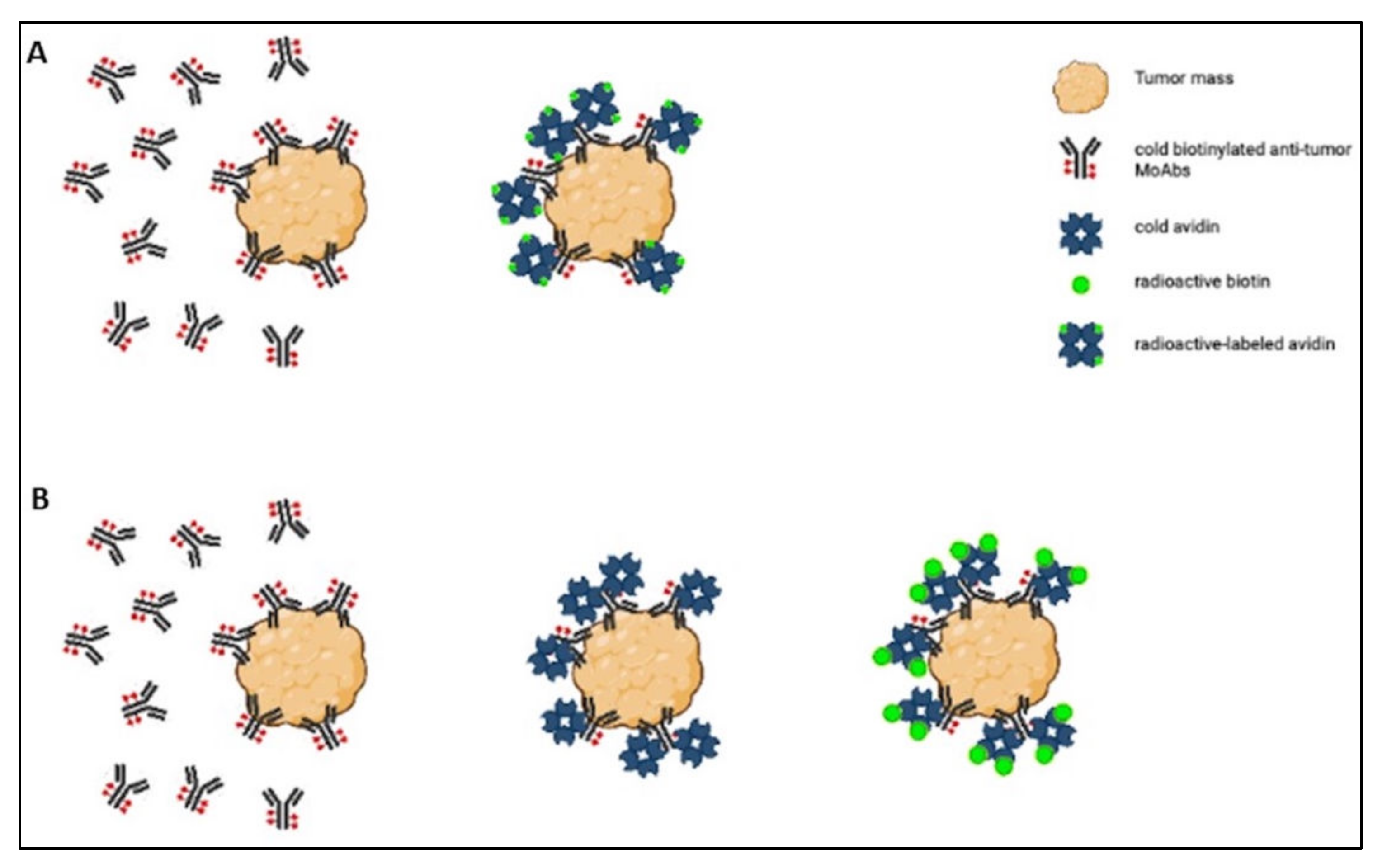
Figure 1. Scheme of the two-step (A) and three-step (B) protocols.
In the two-step protocol, the tumor is first targeted with cold biotinylated antitumor MoAbs, and then radioactive-labeled avidin is administered [27]. The three-step protocol involves (1) tumor pretargeting with cold biotinylated antitumor antibodies, (2) administering cold avidin to remove circulating biotinylated antibodies and ensure avidination of the biotinylated tumor-bound antibodies, and (3) labeling the tumor with radioactive biotin derivatives [28]. Both protocols have been tested in preclinical and clinical settings, but only the clinical studies are considered. Here are nine articles that could be divided according to the type of tumor treated: three articles concerning various types of tumors, three articles concerning gliomas, and three articles dealing with breast cancer.
Paganelli and coworkers [28] first tested the feasibility of the three-step protocol in 20 patients with tumors expressing carcinoembryonic antigen (CEA). Their preliminary study, conducted with 111In, revealed the advantages and disadvantages of the technique compared with the direct administration of radiolabeled MoAbs. The advantages included a drastically reduced background radioactivity, a well-preserved MoAb immunoreactivity (as autoradiolysis-induced damage to the MoAbs was avoided), and signal amplification. The main disadvantages were the need for repeated injections and the immunogenicity of avidin.
Some years later, Cremonesi and coworkers [29] described the pharmacokinetic properties of the three-step protocol. The organs receiving the highest doses of radioactivity were the kidneys, liver, and urinary bladder, but the levels of renal, hepatic, or hematological toxicity were low.
Paganelli and coworkers [27] also examined the application of the two-step protocol in the treatment of 15 patients with ovarian carcinoma. Here again, the high tumor-to-normal tissue ratio was highlighted as the main advantage of the method, and the repeated injections and use of streptavidin (an avidin analog) were identified as the main drawbacks.
After these first promising reports, the use of pretargeting strategies in tumor therapy spread and came to be applied to the treatment of malignant high-grade gliomas. In a phase I/II study, Paganelli and coworkers [30] used a three-step protocol to deliver 15 times more radioactivity to the sites of brain tumors than to critical organs (e.g., liver and kidneys). The treatment’s toxicity was consequently acceptable, with most of the activity not bound to the tumor eliminated in the first 24 h. The therapeutic benefit was evident in most patients (the tumor progressed no further in 52% of cases and shrank significantly in 25%), and the response persisted for more than 1 year in some patients. In their study, the only major drawback was again immunogenicity due to the administration of streptavidin. To solve this problem, the authors recommended using avidin modified with polyethylene glycol (PEG) molecules instead of streptavidin, as PEG can hide avidin from the immune system. Grana and coworkers [31] also tested a three-step protocol in the treatment of malignant gliomas, with promising results. The authors suggested that associating this technique with surgery, radiotherapy, and chemotherapy might increase the life expectancy of patients with high-grade gliomas. When Paganelli and coworkers [32] applied the three-step protocol in the locoregional treatment of patients with high-grade gliomas, they reported an objective therapeutic response in many patients, along with an encouraging median overall survival. On the basis of the neurological toxicity observed, they identified 1.11 GBq as the maximum tolerated dose.
All these studies on the avidin–biotin interaction led Paganelli and coworkers to develop a new procedure called IART (intraoperative avidination for radionuclide therapy) for use in the treatment of breast cancer. This procedure consists of two main steps: (1) “avidination” of the anatomical area of the lesion directly after tumor resection; (2) intravenous injection of radiolabeled biotin to target the anatomical area of the tumor 1 day after surgery. Before the radiolabeled biotin is injected, the circulating avidin is removed by injecting an appropriate amount (20 mg) of biotinylated albumin [24][25][33]. The rationale behind this procedure is that the inflammatory reaction after surgery makes the breast tissue a cation exchanger, thus enabling avidin retention at the site affected for several days [24][25][33]. The first studies using IART generated information on the biodistribution of biotin, which was labeled with 111In via the DOTA chelator. The radiolabeled biotin uptake appeared to be fast and stable at the operated tumor site, with a rapid blood and renal clearance, as well as a consequently reduced toxicity. The doses absorbed by the most affected organs (bladder and kidneys) were well below the threshold doses reported in the literature [24][33]. A more recent, phase II study was performed by Paganelli and coworkers [25] to quantify the doses administered with IART. The biologically effective dose (BED) to the tumor bed was 21 Gy when a fixed activity of 3.7 GBq of 90Y-DOTA-biotin was injected. The authors judged that IART can consequently be considered as a boost to tumor treatment, especially in association with EBRT. They concluded that this technique may be applicable not only to any breast cancer amenable to conservative surgery, as well as to many other solid tumors such as those involving the bladder, prostate, and brain [24][25][33].
4. Docking
Another possible strategy for performing an accurate TRT is to use a ligand with a very high affinity for the target. To ensure the strongest and most specific interaction, the ligand can be designed ad hoc, according to the structure of other known ligands or to the ligand’s interaction with the receptor. One way to ascertain whether the ligand thus designed is active is to use docking, a virtual simulation of ligand–receptor binding. The simulation of the interaction returns a score that can be used to quantify the ligand’s ability to bind its receptor.
Here are five recent studies on the application of docking in the development of a ligand suitable for radionuclide delivery. In three of the five articles, docking was used to select the ligands with the strongest interaction with the receptor, while it was used to justify the results obtained in the other two articles. Some studies dealt with imaging rather than therapeutic goals (as the use of docking in ligand development is relatively new, there has been too little time to assess all the ligands for therapeutic applications), but introduce them here to better explain the docking method.
Yang and coworkers [34] designed several new ligands for prostate-specific membrane antigen (PSMA) and used docking to identify the most promising among them. They synthesized two series of ligands based on a carbamate structure; one contained the amino-pentanedioic acid (NPA) moiety, while the other contained the oxypentanedioic acid (OPA) moiety. Then, they used docking to test the interaction between the carbamate derivatives and the PSMA. Their results showed that the Lys-OPA carbamates were better ligands than the Lys-NPA carbamates. Two of the former showed a high target-selective uptake in tumor xenografts, and one of the two (4-bromo-2-[18F] fluorobenzoyllysine OPA carbamate) also had a rapid normal organ clearance, making it the most likely candidate for clinical application.
Somatostatin receptor 2 (SSTR2) was the object of efforts to develop new ligands for treating neuroendocrine tumors (NETs). This receptor is the target of the previously mentioned somatostatin analogs DOTA-TOC and DOTA-TATE, but the focus of attention has recently shifted to developing receptor antagonists because they can bind to a larger number of sites, leading to a higher tumor uptake [35]. With this in mind, Behnammanesh and coworkers [35] developed a series of SSTR2 antagonists and labeled them with 177Lu by means of the DOTA chelator. Docking analysis then helped the authors to identify the peptide with the most successful accommodation at the binding site of the receptor (the DOTA-peptide 2, DOTA-p-Cl-Phe-Cyclo(d-Cys-l-BzThi-d-Aph-Lys-Thr-Cys)-d-Tyr-NH2). The same peptide was subsequently synthesized and tested in vitro and in vivo; it showed a good stability, had suitable pharmacokinetic properties, and was able to reveal tumor lesions, making it a promising therapeutic agent for NETs.
This entry is adapted from the peer-reviewed paper 10.3390/cimb44080225
References
- Gill, M.R.; Falzone, N.; Du, Y.; Vallis, K.A. Review targeted radionuclide therapy in combined-modality regimens. Lancet Oncol. 2017, 18, e414–e423.
- Gudkov, S.V.; Shilyagina, N.Y.; Vodeneev, V.A.; Zvyagin, A.V. Targeted radionuclide therapy of human tumors. Int. J. Mol. Sci. 2015, 17, 33.
- Artigas, C.; Mileva, M.; Flamen, P.; Karfis, I. Targeted radionuclide therapy: An emerging field in solid tumours. Curr. Opin. 2021, 33, 493–499.
- Peltek, O.O.; Muslimov, A.R.; Zyuzin, M.V.; Timin, A.S. Current outlook on radionuclide delivery systems: From design consideration to translation into clinics. J. Nanobiotechnol. 2019, 17, 90.
- Malcolm, J.; Falzone, N.; Lee, B.Q.; Vallis, K.A. Targeted radionuclide therapy: New advances for improvement of patient management and response. Cancers 2019, 11, 268.
- Li, S.; Goins, B.; Zhang, L.; Bao, A. Novel multifunctional theranostic liposome drug delivery system: Construction, characterization, and multimodality MR, near-infrared fluorescent, and nuclear imaging. Bioconjug. Chem. 2012, 23, 1322–1332.
- Yang, F.; Wang, H.; Liu, R.; Teng, M.; Li, J.; Lu, M. Pharmacokinetic analysis of 111 In-labeled liposomal doxorubicin in murine glioblastoma after blood-brain barrier disruption by focused ultrasound. PLoS ONE 2012, 7, e45468.
- Jensen, G.M.; Bunch, T.H. Conventional liposome performance and evaluation: Lessons from the development of Vescan. J. Liposome Res. 2007, 17, 121–137.
- Jensen, G.M.; Hodgson, D.F. Opportunities and challenges in commercial pharmaceutical liposome applications. Adv. Drug Deliv. Rev. 2020, 154–155, 2–12.
- Du, Y.; Liang, X.; Li, Y.; Sun, T.; Jin, Z.; Xue, H.; Tian, J. Nuclear and fluorescent labeled PD-1-Liposome-DOX-64 Cu/IRDye800CW allows improved breast tumor targeted imaging and therapy. Mol. Pharm. 2017, 14, 3978–3986.
- Silva, J.O.; Fernandes, R.S.; Lopes, S.C.A.; Cardoso, V.N.; Leite, E.A.; Cassali, G.D.; Marzola, M.C.; Rubello, D.; Oliveira, M.C.; Luis, A.; et al. pH-sensitive, long-circulating liposomes as an alternative tool to deliver doxorubicin into tumors: A feasibility animal study. Mol. Imaging Biol. 2016, 18, 898–904.
- Monteiro, L.O.F.; Fernandes, R.S.; Oda, C.M.R.; Lopes, S.C.; Townsend, D.M.; Cardoso, V.N.; Oliveira, M.C.; Leite, E.A.; Rubello, D.; Barros, A.L.B. De biomedicine & pharmacotherapy paclitaxel-loaded folate-coated long circulating and pH-sensitive liposomes as a potential drug delivery system: A biodistribution study. Biomed. Pharmacother. 2018, 97, 489–495.
- Srimathveeravalli, G.; Abdel-atti, D.; Carlos, P.; Takaki, H.; Solomon, S.B.; Mulder, W.J.M.; Reiner, T. Reversible electroporation—mediated liposomal doxorubicin delivery to tumors can be monitored with 89 Zr-Labeled reporter nanoparticles. Mol. Imaging 2018, 17, 1536012117749726.
- Luo, D.; Goel, S.; Liu, H.; Carter, K.A.; Jiang, D.; Geng, J.; Kutyre, C.J.; Engle, J.W.; Huang, W.; Shao, S.; et al. Intrabilayer 64 Cu labeling of photoactivatable, doxorubicin-loaded stealth liposomes. ACS Nano 2017, 11, 12482–12491.
- Lamichhane, N.; Dewkar, G.K.; Sundaresan, G.; Mahon, R.N.; Zweit, J. -fluorinated carboplatin and -liposome for image-guided drug delivery. Int. J. Mol. Sci. 2017, 107, 1079.
- Gawne, P.; Man, F.; Fonslet, J.; Radia, R.; Bordoloi, J.; Long, N.; Rosales, R.T.M. De and liposomal nanomedicine PET imaging using. Dalt. Trans. 2018, 47, 9283–9293.
- Lin, Y.; Kao, H.; Li, J.; Hwang, J.; Tseng, Y.; Lin, W.; Lin, M.; Ting, G.; Wang, H. Tumor burden talks in cancer treatment with PEGylated liposomal drugs. PLoS ONE 2013, 8, e63078.
- Edmonds, S.; Volpe, A.; Shmeeda, H.; Parente-pereira, A.C.; Radia, R.; Bagun, J.; Szanda, I.; Severin, G.W.; Livieratos, L.; Blower, P.J.; et al. Exploiting the metal-chelating properties of the drug cargo for in vivo positron emission tomography imaging of liposomal nanomedicines. ACS Nano 2016, 10, 10294–10307.
- Lee, H.; Zheng, J.; Gaddy, D.; Orcutt, K.D.; Leonard, S.; Geretti, E.; Hesterman, J.; Harwell, C.; Hoppin, J.; Jaffray, D.A.; et al. A gradient-loadable 64Cu-chelator for quantifying tumor deposition kinetics of nanoliposomal therapeutics by positron emission tomography. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 155–165.
- Ito, K.; Hamamichi, S.; Asano, M.; Hori, Y.; Matsui, J.; Iwata, M.; Funahashi, Y.; Umeda, I.O.; Fujii, H. Radiolabeled liposome imaging determines an indication for liposomal anticancer agent in ovarian cancer mouse xenograft models. Cancer Sci. 2015, 107, 60–67.
- Arrieta, Ó.; Medina, L.A.; Estrada-Lobato, E.; Hernández-Pedro, N.; Villanueva-Rodríguez, G.; Martínez-Barrera, L.; MacEdo, E.O.; López-Rodríguez, V.; Motola-Kuba, D.; Corona-Cruz, J.F. First-line chemotherapy with liposomal doxorubicin plus cisplatin for patients with advanced malignant pleural mesothelioma: Phase II trial. Br. J. Cancer 2012, 106, 1027–1032.
- Arrieta, O.; Medina, L.A.; Estrada-Lobato, E.; Ramírez-Tirado, L.A.; Mendoza-García, V.O.; De La Garza-Salazar, J. High liposomal doxorubicin tumour tissue distribution, as determined by radiopharmaceutical labelling with 99mTc-LD, is associated with the response and survival of patients with unresectable pleural mesothelioma treated with a combination of liposomal do. Cancer Chemother. Pharmacol. 2014, 74, 211–215.
- Lee, H.; Shields, A.F.; Siegel, B.A.; Miller, K.D.; Krop, I.; Ma, C.X.; Lorusso, P.M.; Munster, P.N.; Campbell, K.; Gaddy, D.F.; et al. 64Cu-MM-302 positron emission tomography quantifies variability of enhanced permeability and retention of nanoparticles in relation to treatment response in patients with metastatic breast cancer. Clin. Cancer Res. 2017, 23, 4190–4202.
- Paganelli, G.; Ferrari, M.; Cremonesi, M.; De Cicco, C.; Galimberti, V.; Luini, A.; Veronesi, P.; Fiorenza, M.; Carminati, P.; Zanna, C.; et al. IART®: Intraoperative avidination for radionuclide treatment. A new way of partial breast irradiation. Breast 2007, 16, 17–26.
- Paganelli, G.; De Cicco, C.; Ferrari, M.E.; Carbone, G.; Pagani, G.; Leonardi, M.C.; Cremonesi, M.; Ferrari, A.; Pacifici, M.; Di Dia, A.; et al. Intraoperative avidination for radionuclide treatment as a radiotherapy boost in breast cancer: Results of a phase II study with 90Y-labeled biotin. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 203–211.
- Chinol, M.; De Cobelli, O.; Trifirò, G.; Scardino, E.; Bartolomei, M.; Verweij, F.; Papi, S.; Matei, D.V.; Paganelli, G. Localization of avidin in superficial bladder cancer: A potentially new approach for radionuclide therapy. Eur. Urol. 2003, 44, 556–559.
- Paganelli, G.; Belloni, C.; Magnani, P.; Zito, F.; Pasini, A.; Sassi, I.; Meroni, M.; Mariani, M.; Vignali, M.; Siccardi, A.G.; et al. Two-step tumour targetting in ovarian cancer patients using biotinylated monoclonal antibodies and radioactive streptavidin. Eur. J. Nucl. Med. 1992, 19, 322–329.
- Paganelli, G.; Malcovati, M.; Siccardi, A.G.; Villa, E.; Sudati, F.; Rossetti, C.; Fazio, F. Three-step monoclonal antibody tumor targeting in carcinoembryonic antigenpositive patients. Cancer Res. 1991, 51, 5960–5966.
- Cremonesi, M.; Ferrari, M.; Chinol, M.; Stabin, M.G.; Grana, C.; Prisco, G.; Robertson, C.; Tosi, G.; Paganelli, G. Original article Three-step radioimmunotherapy with yttrium-90 biotin: Dosimetry and pharmacokinetics in cancer patients. Eur. J. Nucl. Med. 1999, 26, 110–120.
- Paganelli, G.; Grana, C.; Chinol, M.; Cremonesi, M.; De Cicco, C.; De Braud, F.; Robertson, C.; Zurrida, S.; Casadio, C.; Zoboli, S.; et al. Antibody-guided three-step therapy for high grade glioma with yttrium-90 biotin. Eur. J. Nucl. Med. 1999, 26, 348–357.
- Grana, C.; Chinol, M.; Robertson, C.; Mazzetta, C.; Bartolomei, M.; De Cicco, C.; Fiorenza, M.; Gatti, M.; Caliceti, P.; Paganelli, G. Pretargeted adjuvant radioimmunotherapy with Yttrium-90-biotin in malignant glioma patients: A pilot study. Br. J. Cancer 2002, 86, 207–212.
- Paganelli, G.; Bartolomei, M.; Ferrari, M.; Cremonesi, M.; Broggi, G.; Maira, G.; Sturiale, C.; Grana, C.; Prisco, G.; Gatti, M.; et al. Pre-targeted locoregional radioimmunotheraphy with 90Y-biotin in glioma patients: Phase I study and preliminary therapeutic results. Cancer Biother. Radiopharm. 2001, 16, 227–235.
- Paganelli, G.; Ferrari, M.; Ravasi, L.; Cremonesi, M.; De Cicco, C.; Galimberti, V.; Sivolapenko, G.; Luini, A.; De Santis, R.; Travaini, L.L.; et al. Intraoperative avidination for radionuclide therapy: A prospective new development to accelerate radiotherapy in breast cancer. Clin. Cancer Res. 2007, 13, 5646–5652.
- Yang, X.; Mease, R.C.; Pullambhatla, M.; Lisok, A.; Chen, Y.; Foss, C.A.; Wang, Y.; Shallal, H.; Edelman, H.; Hoye, A.T.; et al. Fluorobenzoyllysinepentanedioic acid carbamates: New scaffolds for positron emission tomography (PET) imaging of prostate-speci fi c membrane antigen (PSMA). J. Med. Chem. 2016, 59, 206–218.
- Behnammanesh, H.; Jokar, S.; Erfani, M.; Geramifar, P. Design, preparation and biological evaluation of a 177Lu-labeled somatostatin receptor antagonist for targeted therapy of neuroendocrine tumors. Bioorg. Chem. 2020, 94, 103381.
This entry is offline, you can click here to edit this entry!