Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
The inflammatory process plays a significant role in the development of colon cancer (CRC). Intestinal cytokine networks are critical mediators of tissue homeostasis and inflammation but also impact carcinogenesis at all stages of the disease.
- inflammation
- cytokines
- cancer
- colorectal
1. Introduction
Colorectal cancer (CRC) is the third most common and fourth most deadly malignancy worldwide, characterized by a sequential accumulation of multiple genetic aberrations [1]. Epithelial inflammation has been recently deemed as one of the hallmarks of CRC pathogenesis factors, just next to genetic abnormalities. Inflammation impacts all stages of carcinogenesis, including initiation, promotion, and progression [2,3]. Among other mediators of inflammation, cytokines seem to have a unique but complicated role in driving or preventing malignant transformation. The harmony between proinflammatory and anti-inflammatory factors is crucial to maintaining homeostasis. Wherever the balance is shifted towards either side, CRC initiation may occur [4].
In the context of coexistent inflammation, colorectal cancers can be classified into two main subtypes, namely sporadic or colitis-associated cancer. Sporadic cancer arises without known germline mutations, family history of cancer, or inflammatory bowel disease [5]. It is the most common type seen in practice and accounts for more than 60% of CRC cases. Usually, the progression from adenoma to carcinoma takes over 1–2 decades and presents mostly after 60 years of age [1]. Colitis-associated cancer (CAC) is a type of colorectal cancer whose pathogenesis is associated with long-term inflammation. Its occurrence is preceded by clinically detectable inflammatory bowel diseases (IBD), while the risk of CAC correlates with the time and severity of the active disease. CAC is characterized by the formation of polyploid, diffuse or multifocal, and invasive lesions compared to sporadic CRC [6,7]. The preexisting inflammatory processes seem to affect carcinogenesis by causing cellular stress, limiting immune surveillance, rendering the DNA damage response, and accelerating the acquisition of genomic alterations. Both sporadic and colitis-associated cancers are the most frequently characterized by increasing microsomal and chromosomal instability; however, they differ in timing and frequency of specific alterations [3,8,9,10]. Some of them seem pivotal in the early steps of cancerogenesis. For example, the mutation of p53, found in up to 85% of CACs, occurs as one of the first events in the CAC cascade and is more frequent there than in sporadic cancers [11] (Figure 1).
Sporadic cancer and CAC show a similar density of somatic mutations, contradicting the assumption that inflammation outright increases the number of mutations in CAC when compared to sporadic cancers [12]. Nevertheless, CACs harbor some unique genetic alterations. While the frequency of PIK3CA, BRAF, and SMAD4 mutations was similar in both types of cancer, changes affecting genes responsible for cell mobility, cytoskeleton remodeling, and those required for p53 transcription were more frequent in CACs. Noteworthy, Robles et al. detected the amplification of a region-encoding suppressor of cytokine signaling 1 (SOCS1) in CAC tumors. Since SOCS1 downregulates cytokine signaling, including the antitumor activity of IFN-γ and IL-27, its amplification may limit tumor immunosurveillance and enhance inflammation-driven CAC [11,12].
The uniqueness of the genomic landscape of CAC sheds some light on the link between Crohn’s disease (CD), ulcerative colitis (CU), and colorectal cancer. The cumulative risk of CAC reaches up to 8% in patients with CD and CU at 20 years of disease, while CAC causes the death of 15% of IBD patients [13]. Although those diseases differ in cytokine profile, both are associated with significant dysregulation of cytokine transmission. In CD, the predominant type 1 helper T cells (Th1) induce mostly IL-12, IFN-γ, and TNF-α signaling, but in CU, the Th2 cytokines, such as IL-5 and IL-13, play a major role [14]. Furthermore, both CD and CU have distinctive cytokine profiles. While both diseases show frequent downregulation of IL-7, as well as the overexpression of IL-6, IL-12, IL-18, IL-21, IL-27, and IL-34, the overexpression of IL-17, IL-23, and IL-32 seems more specific for CD [15,16,17,18]. On the contrary, higher levels of 5, IL-13, IL-15, IL-23R, and IL-33 were prevalent in CU [19,20].
That imbalance can disturb the local cytokine network, lead to prolonged epithelial stress, and cause tissue injury. In settings of reduced apoptotic stimuli and enhanced proliferation, the risk of genetic error increases, leading to the initiation of carcinogenesis [2,6]. The disruption in the cytokine-mediated crosstalk between immune and epithelial cells in the large bowel seems to be a major factor driving chronic inflammation and leading to the initiation and progression of large bowel cancers [21]. Epidemic studies seem to partly support the hypothesis that chronic inflammation accelerates the formation of cancer because colitis-associated cancers develop in younger patients, are often localized in the right colon, and are characterized by shorter overall survival [22]. Therefore, clarifying the role of underlying cytokine pathways and the effects of their modulation may be an important step to improve the effectiveness of cancer immunotherapy.
2. Inflammation in the Pathways of Sporadic and Colitis-Associated Colorectal Carcinogenesis
A growing number of studies report that inflammation can influence the dynamics and aggressiveness of still-forming malignancy [2,10,21]. Canonically, there are three main pathways of genetic instability and two morphological sequences of CRC carcinogenesis, which differ in pathogenesis and dynamics [10,23,24] (Figure 1). Around 60–85% of CRCs develop through the “classic” adenoma-carcinoma pathway, which may be a physical manifestation of either the chromosomal (CIN) or microsatellite instability (MSI). While the first step in both is the loss of APC function, in the CIN pathways it is usually the sole mutation, which leads to the formation of early adenoma [23]. The following activating mutation of KRAS, Wnt, or other oncogenes, or the loss of 18q LOH, leads to the appearance of a late adenoma stage, which then transforms into adenocarcinoma as a result of the loss of p53 [10]. In the case of the MSI pathway, adenoma is the result of a simultaneous loss of APC and DNA mismatch repair genes (MMR) failure. Hence, a subsequent BRAF, Bax, or TGFβR mutation pushes the neoplasm to the carcinoma stage [25].
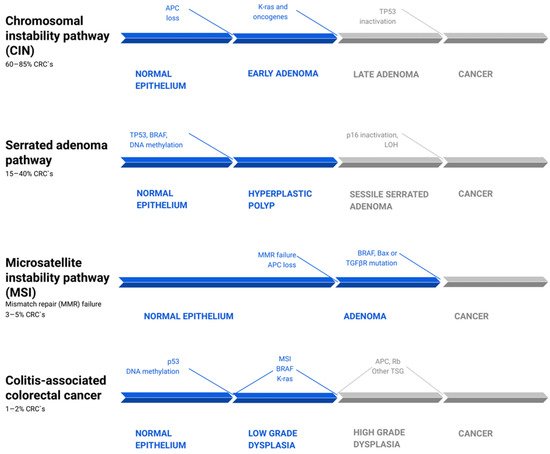
Figure 1. Common pathways of colorectal cancer pathogenesis [10,23,25,26,27]. There are four parallel pathways involved in CRC progression. The chromosomal instability pathway (CIN) accounts for 60–85% of CRC. Mutations, such as APC loss, activation of K-ras or other oncogenes, and then Tp53 inactivation, drive clonal cell growth and ultimately the formation of invasive cancer. The serrated adenoma pathway is responsible for 15–40% of CRC. The occurrence of a BRAF mutation and methylation of cell cycle controlling leads to uncontrolled proliferation of tumor cells. Subsequent methylation of other genes, such as tp53 and p16, promotes the evolution of CRC. Microsatellite instability pathway (MSI) failure leads to 3–5% of CRCs and is driven by simultaneous loss of APC and DNA mismatch repair genes (MMR) failure. The following BRAF, Bax, or TGFβR mutation pushes the tumors from adenoma to CRC. Colitis-associated colorectal cancer accounts for about 1–2% of all CRCs. Its mechanism is similar to that in the pathogenesis of sporadic cancer, including p53, DNA methylation, MSI, BRAF, and K-ras mutations. However, they differ in timing and frequency of specific alterations. These mutations lead to low-grade dysplasia, which, after mutations of APC, Rb, and other TSG, progress to high-grade dysplasia and subsequently to cancer. CRC—colorectal carcinoma; LOH—loss of heterozygosity; TSG—tumor suppressor genes.
The “alternative” serrated pathway, in which 15–40% of CRC develops, is associated with the BRAF mutation and excessive methylation of cell cycle control proteins, which causes the occurrence of serrated adenoma. The pathogenesis of the remaining CRC cases is still not fully explained [28]. In contrast to the pathogenetic pathway of sporadic cancers, colitis-related cancers are better characterized by a “dysplasia-carcinoma” model [8]. In this model, most mutations, including APC and KRAS, occur less often than in sporadic CRC and at later stages of tumor evolution, where they promote cytokine secretion, tumor growth, and angiogenesis via the NF-κB, IL-6/STAT3, or IL-23/Th17 pathways [29,30,31]. APC mutations occur less often and later in CACs than in sporadic cancers, but they often express high levels of β-catenin. Given that APC encodes a negative regulator of β-catenin, suppressing the tumorigenic activity of the Wnt/β-catenin pathway, there is a possibility that chronic inflammation and tissue injury play a major role in the downstream deregulation of Wnt/β-catenin signaling. Those changes may occur even in the absence of APC mutations (Figure 1) [11,12,32].
Most adenomas occur through Wnt-driven transformation of stem cells, while serrated polyps originate from differentiated cells undergoing gastric metaplasia [33]. Importantly, genetically unstable CRCs, outside of the metaplastic lesions, contain adjacent non-metaplastic regions, in which immune cells become exhausted and tumor cells acquire stem-like properties [33]. TGF-β, PI3K, and Wnt signaling alterations are the most frequent in sessile serrated adenoma [34].
Reportedly, mutant p53 can drive CAC carcinogenesis by prolonging TNF-α-induced NF-κB activation in cultured cells and increasing IL-6 and IL-8 levels [35]. Inflammation may not be a decisive factor in CRC initiation, and CAC risk correlates with the time and severity of active disease [7]. In the presence of DNA damage response failure, mismatch repair defects, and oncogene-induced replication stress, the coexistent inflammatory process can exacerbate the genetic burden and accelerate tumorigenesis through NF-κB-mediated cytokine production [9]. The semi-local inflammation promotes the accumulation of chemokines, immune cells, stromal cells, and extracellular matrix proteins, which under favorable conditions provide the foundation for the development of the tumor-supportive microenvironment [36].
Interleukin-1β, IL-4, and TNF-α are overexpressed in early events of CRC development, such as hyperplastic polyps, adenomas, and serrated adenomas. However, in adenocarcinomas, IL-4 levels are not elevated compared with normal mucosa [37]. The expression of IL-4, TNF-α, COX-2, and IL-1β is higher in serrated adenomas than in adenomas, but the expression of IL-10 shows the opposite trend. Those results suggest that inflammation can be of greater importance in the serrated pathway [38].
3. Cytokines, Tumor Microenvironment, and Epithelial–Mesenchymal Transition
In recent years, we have made significant progress to understand the pathophysiology of the tumor microenvironment (TME) and how the initiation of epithelial–mesenchymal transition (EMT) enables cancer cells’ migration and metastasis. Although new reports are still being published, it is already known that interleukins can mediate the crosstalk between cancer cells and the adjacent tissues, impacting carcinogenesis [39].
The TME consists of the physical and cellular vicinity of the primary tumor and includes the tumor stroma, tumor-associated cells, immune cells, endothelial cells, macrophages, vessels, and various components of the extracellular matrix (ECM). The immune components of TME are called the tumor immune microenvironment (TIME) and can modulate tumor evolution [39,40]. TIME is composed mostly of myeloid-derived stem cells and regulatory T cells, which mediate local immunosuppression, and cytotoxic T lymphocytes (CTLs), Th2 cells, and macrophages M2, which possess very restricted antitumor activity [41,42]. During the progression of the disease, cancer cells change the properties of surrounding cells, forcing them to produce growth factors and proinflammatory cytokines, such as TNF-α, IL-1β, and TGF-β, that accelerate the acquisition of a mesenchymal-like phenotype during EMT [36].
EMT is a mechanism that allows the cancer cells to transgress their epithelial features and acquire mesenchymal-like properties [43]. The induction of EMT can be triggered by transcription factors, such as ZEB1, whose expression increases with cancer stages and is associated with aggressive disease and poor prognosis in colon cancer [44]. While EMT can occur during wound healing or fibrosis, it is also assumed to be a key player in cancer. As a result of cancer–stromal crosstalk, stationary epithelial cells lose the intracellular adhesive properties, gain spindle cell-like mobility, and become able to migrate from primary tumors [36].
Once the cancer cells exit the bloodstream and form micrometastases, a reverse process, called mesenchymal–epithelial transition, enables them to form metastatic lesions [45]. Tumor-associated macrophages (TAM), myeloid-derived stem cells (MDSC), and lymphocytes Th1/Tc infiltrating the TME influence the epithelial cells undergoing TME through the secretion of IL-1β, IL6, IL8, IL10, IL17, TGF-β, or TNFα [46]. Under the influence of IL-1β, IL-8, and TGFβ, which can affect Wnt/β-catenin signaling, cancer cells can gain stem-like qualities and form a subpopulation of cancer stem cells capable of self-renewal and recreation of the entire tumor population [47]. Simultaneously, the infiltration of suppressive immune cells into the TME increases, and the immunophenotype of local cells changes, leading to the failure of immune surveillance and cancer immune escape [36,39,45,47]. Hence, tumor-associated signaling is essential in understanding the pathogenesis of CRC and in applying potential therapeutic approaches.
4. The Mediatory Role of Interleukins in Colorectal Carcinogenesis
Interleukins have gained the most attention due to their potential role in CRC pathogenesis and promising results of clinical trials. They form several families and subfamilies, which have distinct immunomodulatory effects and can cause CRC promotion, tumor growth, and metastasis, or prevent them [48,49]. Malignant transformation is associated with the pro-tumorigenic and anti-tumorigenic interleukins. The harmony between proinflammatory and anti-inflammatory factors is crucial to maintaining homeostasis (Figure 2).
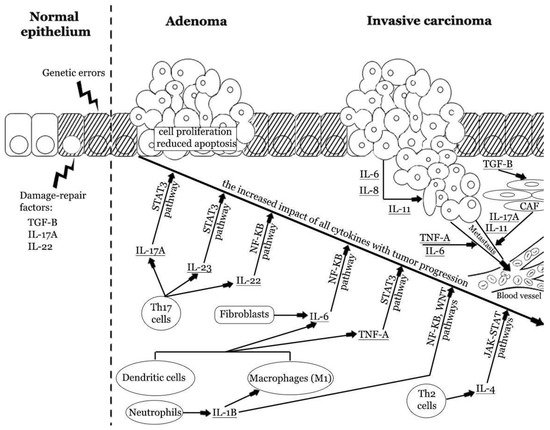
Figure 2. The impact of pro-tumorigenic cytokines in colorectal carcinogenesis. Repetitive colorectal mucosal damage may induce chronic immune system activation and injury of epithelial cells. These processes stimulate cellular augmented proliferation and regeneration, which in turn may result in the accumulation of genetic errors. Inflammatory cytokines may promote tumor formation and enhance progression from adenoma to invasive carcinoma. Cytokines produced by innate and adaptive immune cells and fibroblasts promote cellular proliferation and reduce apoptosis. The latest reports indicate the strong role of IL-17A, IL-6, and TNF-α. These cytokines have an increased impact at different stages of CRC progression. Cancer cells can also produce some cytokines and enhance the vicious cycle of inflammatory response. Finally, cytokines can induce angiogenesis, stromal reorganization, suppression of antitumor immunity, and metastasis [2,29,41,50,51,52,53]. IL—interleukin; Th—T helper cell; TNF-A—tumor necrosis factor α; TGF-β—tissue growth factor β.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10071670
This entry is offline, you can click here to edit this entry!