Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Secondary lymphedema is a common complication of lymph node dissection or radiation therapy for cancer treatment. Conventional therapies such as compression sleeve therapy, complete decongestive physiotherapy, and surgical therapies decrease edema; they are not curative because they cannot modulate the pathophysiology of lymphedema.
- lymphedema
- CD4+ T cell
- mesenchymal stem/stromal cell
1. Introduction
Lymphedema is caused by a dysfunction of the lymphatic system, resulting in localized interstitial fluid retention and tissue swelling; it is classified as primary or secondary. Primary lymphedema develops due to inherited hypoplasia/dysplasia or dysfunction of lymphatic vessels because of some intrinsic factors such as genetic mutations in the signaling pathway for vascular endothelial growth factor C (VEGF-C), while secondary lymphedema is caused by a dysfunction of the lymphatic vascular system due to trauma or parasitic infection [1][2][3]. Although the incidence of secondary lymphedema has been declining due to advances in surgery, this iatrogenic disorder has a strong negative impact on physical and mental quality of life (QOL) [4]. Additionally, radiation therapy (RT) increases the risk of lymphedema in the upper and lower limbs; for instance, in breast cancer patients, the risk of lymphedema is five times higher with postoperative RT than with axillary lymph node dissection alone [5]. Patients with lymphedema typically present with symptoms such as altered mechanical properties and sensitivity of the skin, increased susceptibility to systemic and local infections, decreased function of the affected upper or lower limb, and chronic pain and discomfort [3][6]. In addition, patients may have problems with body image and social acceptability and exhibit low self-esteem [6]. The protein-rich fluid accumulated in the interstitial space induces the migration of CD4+ T-helper (Th) cells, low-grade inflammation, remodeling of extracellular matrix, hyperkeratosis, adipose deposition, and fibrosis [6][7][8][9][10][11]. These changes in the edematous limb exacerbate lymphatic dysfunction, resulting in clinical manifestations of lymphedema.
No curative therapy for lymphedema has been established so far. While conservative therapies (such as manual lymphatic drainage, complete decongestive physiotherapy, compression sleeve therapy, exercise, and weight reduction) decrease edema temporarily, they cannot modulate the pathophysiology of lymphedema. Therefore, it is difficult to maintain their therapeutic efficacy over a lifetime [12][13][14][15][16][17]. Surgical interventions such as lymphovenous anastomosis/bypass or vascularized lymph node transfer (VLNT) are effective in early-stage lymphedema; however, they are ineffective in chronic lymphedema with fibrosis due to lymphatic dysfunction in the edematous region [18][19][20][21][22]. Recently, pharmacotherapy and cell-based therapy have been developed to treat lymphedema by promoting lymphangiogenesis, improving lymphatic function, and suppressing fibrosis and inflammatory responses. Several studies focus on the migration and accumulation of CD4+ T cells in the edematous region as a new target to treat lymphedema [23][24][25][26][27][28]. Mesenchymal stem/stromal cells (MSCs) exert anti-inflammatory, anti-fibrosis, antioxidant stress, and immunomodulatory effects and are hence used in studies to establish cell-based therapy to treat wounds [29], inflammatory bowel diseases [30], diabetes mellitus [31], psoriasis [32][33], and graft-versus-host disease [34]; they are useful since they promote lymphangiogenesis in lymphedema animal models [35][36][37][38][39][40][41][42][43][44][45].
2. Pathophysiology of Secondary Lymphedema
In most regions of the body, lymph flows against a hydrostatic pressure gradient created by extrinsic and intrinsic pump forces arising from the surrounding skeletal muscle and/or lymphatic collecting vessel network [46]. In lymphedema, fluid stasis is caused by lymphatic pump dysfunction due to contractile dysfunction, chronic inflammation, fibrosis, abnormal lymphangiogenesis, barrier dysfunction, or valve defects [46][47]. The protein-rich fluid that accumulates triggers an inflammatory response and exacerbates lymphedema. Although details of the mechanisms remain unclear, the activation of dendritic cells (DCs) and subsequent activation of CD4+ T cells, especially Th2 cell maturation, have been hypothesized as key factors in the inflammatory response (Figure 1) [11][48].
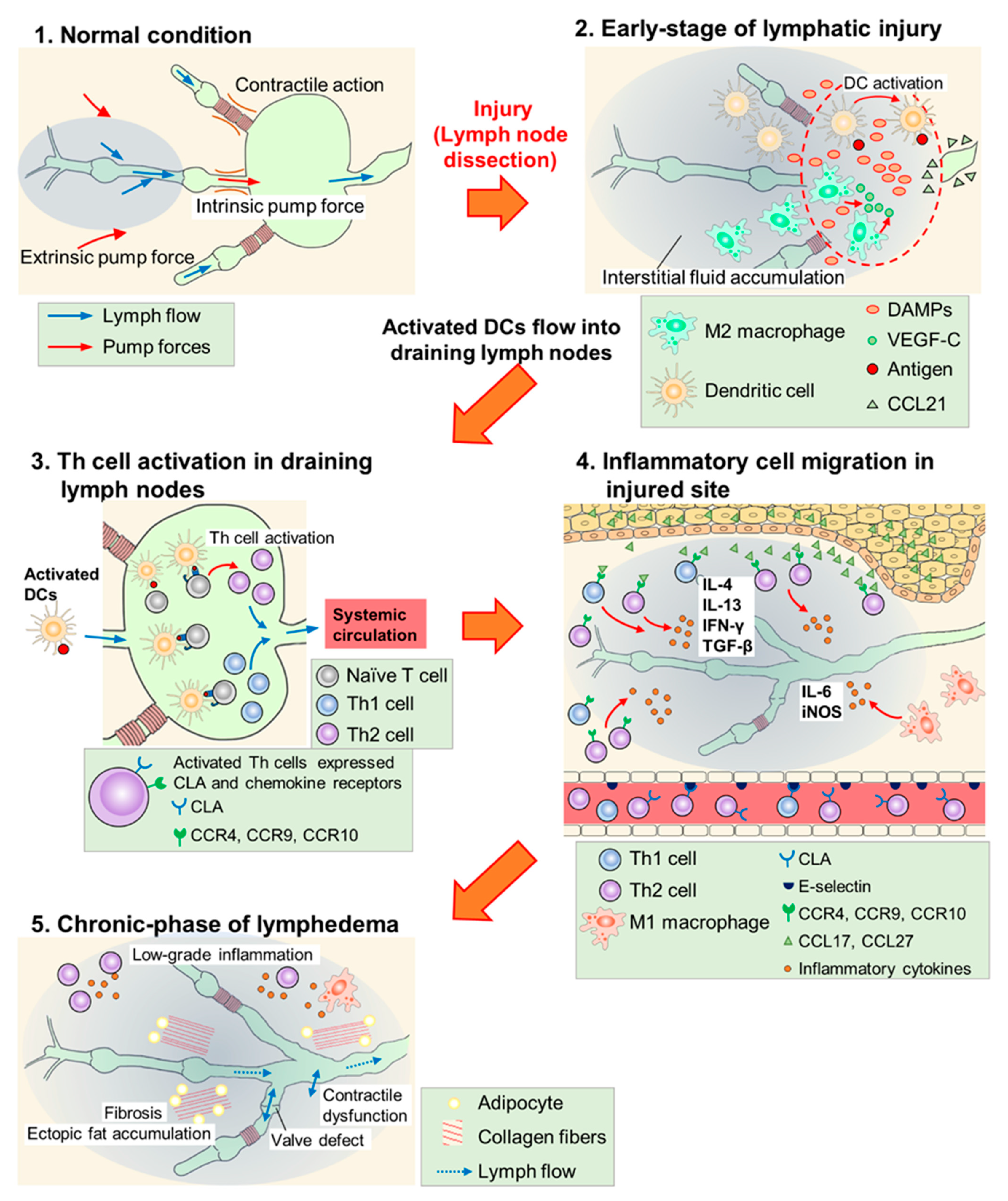
Figure 1. Scheme of lymphedema development after lymph node dissection. (1) In normal conditions, lymph flow is generated due to intrinsic pump force by a lymphatic collecting vessel network and extrinsic pump force by surrounding skeletal muscles. (2) In the early phase of lymphatic injury, damage-associated molecular patterns (DAMPs) are released from injured cells and these molecules promote lymphangiogenesis. M2 macrophages secrete vascular endothelial growth factor C (VEGF-C) and serve as lymphatic endothelial cell (LEC) progenitors. Dendritic cells (DCs) are activated at the injured site, and invade into lymphatic vessels along the concentration gradient of the C–C chemokine ligand (CCL) 21 secreted by LECs. (3) Activated DCs flow into draining lymph nodes and activate helper T (Th) cells. Expressions of cutaneous leukocyte antigen (CLA), C–C chemokine receptor (CCR) 4, CCR9, and CCR10 are increased at the surface of activated Th cells. These cells enter systemic circulation. (4) Activated Th cells, guided by adhesion molecules and CCLs, infiltrate the injured site and secrete inflammatory cytokines. M1 macrophages also accumulate at the injured site and cause inflammatory responses. (5) Low-grade inflammatory responses, fibrosis, adipose deposition, and unfunctional lymphangiogenesis (valve defect and contractile dysfunction) occur in the chronic phase of lymphedema. These responses impair lymphatic function and exacerbate lymphedema.
In the early phase (~6 weeks) of lymphatic injury, endogenous danger signals such as high mobility group box 1 (HMGB1) and heat-shock protein 70 are expressed in endothelial cells, adipocytes, and other stromal cells at the injury site [11][26]. These proteins promote lymphangiogenesis via toll-like receptor signaling and the blockade of HMGB1 activity with glycyrrhizin inhibited inflammatory lymphangiogenesis in the mouse tail lymphedema model [11][26][49][50]. Furthermore, macrophages are recruited, and they accumulate in the lymphedematous region, especially in the early phase [51][52], while M2 macrophages secrete VEGF-C to promote superficial lymphangiogenesis [53]. Shimizu et al. reported that bone marrow-derived M2 macrophages may serve as lymphatic endothelial cell (LEC)-progenitors after adipose-derived regenerative cell (ADRC) treatment in mouse tail lymphedema models [38]. Therefore, innate immune responses may promote lymphangiogenesis and suppress the development of lymphedema in the early phase of lymphatic injury.
In this early phase, DCs accumulate in the injured skin and activate acquired immunity [25]. The expression of C–C chemokine receptor (CCR) type 7 increases on the surface of activated DCs, which migrate according to the concentration gradient of C-C chemokine ligand (CCL) type 21 secreted by LECs. Furthermore, after reaching the lymphatic vessels, activated DCs invade them using intercellular adhesion molecule 1 and/or vascular cell adhesion molecule 1 and flow into the draining lymph nodes [54][55][56][57][58]. In lymph nodes, naïve CD4+ T cells are activated by DCs, and increase the expression of cutaneous leukocyte antigen (CLA), CCR4, CCR9, and CCR10. Furthermore, after entering the bloodstream, activated CD4+ T cells infiltrate the edematous region using adhesion molecules such as E-selectin (a CLA ligand) and migrate toward chemokine ligands for CCR4 (CCL17) and CCR 10 (CCL27). The expression of these adhesion molecules and chemokines increases in the vasculature and keratinocytes of lymphedematous tissue, respectively [25][59][60][61][62][63]. Subsequently, inflammatory cytokines such as interferon (IFN)-γ, interleukin (IL)-4, IL-13, and transforming growth factor (TGF)-β (secreted from the activated CD4+ T cells) promote infiltration of the inflammatory cells, exacerbate fibrosis by collagen deposition, and directly inhibit lymphangiogenesis by suppressing the proliferation, differentiation, and migration of LECs [10][24][64][65][66][67][68][69][70]. Therefore, the activation of CD4+ T cells through antigen presentation by DCs is the key process in the development and exacerbation of lymphedema. This hypothesis is supported by studies using the mouse tail lymphedema models, in which CD4 knockout mice were less likely to develop lymphedema [25] and the depletion of regulatory T cells (Tregs) exacerbated lymphedema [23].
M1 macrophages also infiltrate the lymphedematous region and exacerbate lymphedema by inducing adipose deposition and chronic inflammation via IL-6 [71][72]. In addition, M1 macrophages strongly express inducible nitric oxide synthase (iNOS) and disturb nitric oxide homeostasis maintained by endothelial nitric oxide synthase (eNOS), resulting in attenuated lymphatic vessel pumping [25][73]. These reports suggest that the innate immune system, especially M1 macrophages, is involved in aggravating lymphedema as an inflammatory reaction in the chronic phase.
3. Pharmacotherapy for Lymphedema
Doxycycline, ketoprofen, ubenimex, selenium, synbiotic supplement, tacrolimus, anti-IL-4/IL-13 antibody, fingolimod, and TGF-β inhibitors have been studied for their suppression of inflammatory and oxidative stress. The reported pharmacological mechanisms of these agents are summarized in Figure 2.
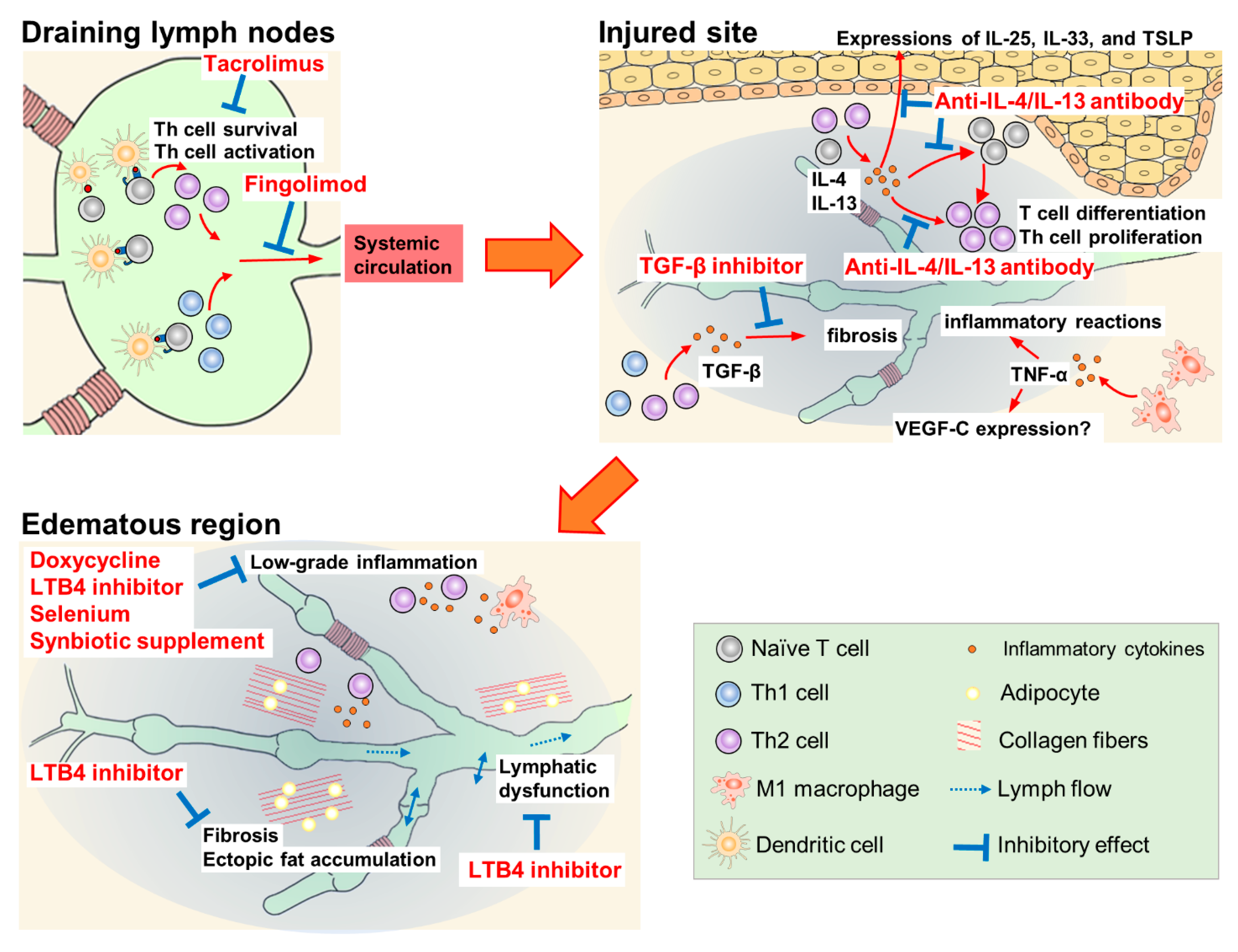
Figure 2. Pharmacological mechanisms of therapeutic agents for lymphedema.
3.1. Doxycycline
Doxycycline, a tetracycline antibiotic, is an anti-Wolbachia drug used for filarial lymphedema. Mand et al. reported that a 6-week course of doxycycline at 200 mg/day improved mild to moderate lymphedema for two years, independent of ongoing filarial infection [74]. The efficacy of doxycycline is probably due to its non-antibiotic effects such as the direct inhibition of inflammation and angiogenesis [75].
3.2. Leukotriene B4 Inhibitors (Ketoprofen, Ubenimex)
Ketoprofen is a non-steroidal anti-inflammatory drug. The efficacy of ketoprofen has been demonstrated in a mouse tail lymphedema model and patients with lymphedema. In a mouse lymphedema model, the subcutaneous injection of ketoprofen decreased tail volume and suppressed histological changes such as epidermal thickening and neutrophil infiltration, while increasing the expression of tumor necrosis factor-α (TNF-α). In contrast, pegsunercept, a modified soluble form of TNF-α receptor R1, increased tail volume, histologically exacerbated the disease, and reduced TNF-α expression. The expression of VEGF-C in this model showed a correlation with TNF-α expression, suggesting that ketoprofen could induce TNF-α-dependent VEGF-C expression followed by lymphangiogenesis [76]. In a clinical study, patients with lymphedema received 75 mg oral ketoprofen three times daily for four months [77]. This treatment significantly improved the histopathology scores (dermal thickness, collagen thickness, intercellular mucin deposits, and perivascular inflammation); however, the volume of the limbs and content of the extracellular fluid were not affected. The mechanism of action of ketoprofen in lymphedema is the inhibition of 5-lipoxygenase activity, which produces leukotriene B4 (LTB4), rather than the inhibition of cyclooxygenase activity [77][78]. In a mouse tail lymphedema model, the intraperitoneal injection of ubenimex (2 mg/kg), a leukotriene A4 hydrolase inhibitor, improved lymphatic collecting vessel pumping, but did not affect the tail volume and leukocyte population in draining lymph nodes [79]. However, the long-term administration of ketoprofen may be inappropriate because of side effects such as acute kidney injury and gastric ulcer due to the non-selective inhibition of physiological cyclooxygenase activity. In contrast, ubenimex inhibits the production of LTB4 selectively. Therefore, ubenimex may be more suitable than ketoprofen for long-term treatment.
3.3. Selenium
The oral and intravenous administration of sodium selenite is effective in the treatment of lymphedema associated with breast cancer and head and neck cancer. In these patients, selenium decreased edema volume and improved the clinical stage of lymphedema. The antioxidant properties of selenium contribute to its efficacy in lymphedema; however, the experimental evidence is unclear [80][81][82]. In a recent study, the intravenous administration of sodium selenite improved lymphoedema and elevated the serum levels of corticosterone, LTB4 dimethylamide (endogenous LTB4 antagonist), and prostaglandin E3 in breast cancer-related lymphedema patients [83]. Elevated levels of these anti-inflammatory substances may be a factor in the therapeutic efficacy of selenium.
3.4. Synbiotic Supplements
Synbiotic supplements, dietary supplements combining probiotics and prebiotics, reduce inflammatory markers such as C-reactive protein (CRP) and TNF-α [84]. In overweight and obese patients with breast cancer-related lymphedema, a 10-week combination of low-calorie diet and synbiotic supplementation resulted in significant reductions in edema volume, serum leptin, and serum inflammatory marker levels (high-sensitivity CRP, IL-1β and TNF-α). However, after adjusting for baseline edema volume, inflammatory marker levels, and body mass index, only serum leptin and TNF-α levels were found to be significantly lower in the synbiotic supplementation group than in the low-calorie diet and placebo capsule group [85]. Although the same research group has reported antioxidant effects and improvement in QOL score with the same dietary combination, the efficacy of synbiotic supplementation for lymphedema has not yet been determined [86][87].
3.5. CD4+ T Cell Suppressants (Tacrolimus, Anti-IL-4/IL-13 Antibodies, Fingolimod)
Treatment with tacrolimus ointment and anti-IL-4/IL-13 antibodies has been shown to suppress the activation and differentiation of CD4+ T cells [24][27][28]. Tacrolimus ointment is used to treat cutaneous inflammatory diseases such as atopic dermatitis. In CD4+ T cells, tacrolimus binds to FK-506-binding protein 12, and the complex inhibits the phosphatase activity of calcineurin, thereby reducing the transcription of IL-2 [88]. CD4+ T cells cannot survive in the presence of tacrolimus because optimal autocrine IL-2 signaling is essential to limit the apoptosis of effector CD4+ T cells and to sustain their transition to and persistence as memory cells [89]. The local administration of tacrolimus in a mouse model with tail lymphedema showed protective and therapeutic efficacy by reducing soft tissue thickness, suppressing inflammatory cell infiltration and inflammatory cytokine expression, and increasing the formation of lymphatic-collecting vessels at the injured site. Furthermore, the recovery of lymphatic functions by tacrolimus, including lymphatic pumping, was observed in a popliteal lymph node dissection model [24].
The inhibition of Th2 differentiation with IL-4- or IL-13-neutralizing antibodies prevents the initiation and progression of lymphedema by inhibiting tissue fibrosis and improving lymphatic function in another mouse tail lymphedema model [27]. Additionally, a report detailed the efficacy of monthly intravenous QBX258 infusion, a combination of two monoclonal antibodies neutralizing IL-4 and IL-13, in eight patients with breast cancer-related lymphedema [28]. Four infusions of QBX258 reduced histological epidermal thickness and suppressed keratinocyte proliferation, type III collagen deposition, mast cell infiltration, and Th2-inducible epithelial-derived cytokine (IL-33, IL-25, and thymic stromal lymphopoietin) expression. QBX258 also improved skin stiffness and patient QOL scores immediately after treatment; however, these improvements returned to baseline four months after the treatment was discontinued. Furthermore, treatment with QBX258 did not decrease the limb volume [28].
Fingolimod (FTY720), a modulator of the sphingosine-1-phosphate receptor, suppresses the emigration of lymphocytes from lymph nodes. In a mouse model with popliteal lymph node dissection, the administration of fingolimod (dissolved in drinking water) from the day of surgery increased CD4+ T cells in inguinal lymph nodes, but decreased them in the skin of the hindlimb [25]. In another mouse tail lymphedema model, fingolimod suppressed the increase in edema volume and fibroadipose thickness from 1 to 6 weeks after lymphatic dissection [25].
3.6. TGF-β Inhibitors (Anti-TGF-β Antibody, Vactosertib, LY-364947)
TGF-β is one of the key mediators in tissue fibrosis; it inhibits functional lymphatic regeneration in the lymphedematous region. The inhibition of TGF-β signaling by monoclonal antibodies or small-molecule drug EW-7197 (vactosertib, an inhibitor of TGF-β receptor type 1) enhanced lymphangiogenesis and lymphatic function by inhibiting fibrosis in a mouse tail lymphedema model [10][90]. Furthermore, LY-364947, a selective inhibitor of TGF-β receptor type 1, markedly suppressed tissue fibrosis and improved lymphatic dysfunction, which was induced by irradiation (with 15 Gy radiation) on the mouse tail [91].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23147614
References
- Grada, A.A.; Phillips, T. Lymphedema: Pathophysiology and clinical manifestations. J. Am. Acad. Dermatol. 2017, 77, 1009–1020.
- Chen, C.; Chiang, N.; Perng, C.; Ma, H.; Lin, C.-H. Review of preclinical and clinical studies of using cell-based therapy for secondary lymphedema. J. Surg. Oncol. 2019, 121, 109–120.
- Brown, S.; Dayan, J.H.; Coriddi, M.; Campbell, A.; Kuonqui, K.; Shin, J.; Park, H.J.; Mehrara, B.J.; Kataru, R.P. Pharmacological Treatment of Secondary Lymphedema. Front. Pharmacol. 2022, 13, 828513.
- Ghezzi, F.; Uccella, S.; Cromi, A.; Bogani, G.; Robba, C.; Serati, M.; Bolis, P. Lymphoceles, Lymphorrhea, and Lymphedema after Laparoscopic and Open Endometrial Cancer Staging. Ann. Surg. Oncol. 2011, 19, 259–267.
- Allam, O.; Park, K.E.; Chandler, L.; Mozaffari, M.A.; Ahmad, M.; Lu, X.; Alperovich, M. The impact of radiation on lymphedema: A review of the literature. Gland Surg. 2020, 9, 596–602.
- Maruccia, M.; Elia, R.; Ciudad, P.; Nacchiero, E.; Nicoli, F.; Vestita, M.; Chen, H.; Giudice, G. Postmastectomy upper limb lymphedema: Combined vascularized lymph node transfer and scar release with fat graft expedites surgical and patients’ related outcomes. A retrospective comparative study. J. Plast. Reconstr. Aesthetic Surg. 2019, 72, 892–901.
- Jensen, M.R.; Simonsen, L.; Karlsmark, T.; Bülow, J. Microvascular filtration is increased in the forearms of patients with breast cancer–related lymphedema. J. Appl. Physiol. 2013, 114, 19–27.
- Zampell, J.C.; Yan, A.; Elhadad, S.; Avraham, T.; Weitman, E.; Mehrara, B.J. CD4+ Cells Regulate Fibrosis and Lymphangiogenesis in Response to Lymphatic Fluid Stasis. PLoS ONE 2012, 7, e49940.
- Avraham, T.; Clavin, N.W.; Daluvoy, S.V.; Fernandez, J.; Soares, M.A.; Cordeiro, A.P.; Mehrara, B.J. Fibrosis Is a Key Inhibitor of Lymphatic Regeneration. Plast. Reconstr. Surg. 2009, 124, 438–450.
- Avraham, T.; Daluvoy, S.; Zampell, J.; Yan, A.; Haviv, Y.S.; Rockson, S.G.; Mehrara, B.J. Blockade of Transforming Growth Factor-β1 Accelerates Lymphatic Regeneration during Wound Repair. Am. J. Pathol. 2010, 177, 3202–3214.
- Kataru, R.P.; Baik, J.E.; Park, H.J.; Wiser, I.; Rehal, S.; Shin, J.Y.; Mehrara, B.J. Regulation of Immune Function by the Lymphatic System in Lymphedema. Front. Immunol. 2019, 10, 470.
- Dayes, I.S.; Whelan, T.J.; Julian, J.A.; Parpia, S.; Pritchard, K.I.; D’Souza, D.P.; Kligman, L.; Reise, D.; Leblanc, L.; McNeely, M.; et al. Randomized Trial of Decongestive Lymphatic Therapy for the Treatment of Lymphedema in Women with Breast Cancer. J. Clin. Oncol. 2013, 31, 3758–3763.
- Schmitz, K.H.; Troxel, A.B.; Dean, L.T.; DeMichele, A.; Brown, J.C.; Sturgeon, K.; Zhang, Z.; Evangelisti, M.; Spinelli, B.; Kallan, M.J.; et al. Effect of Home-Based Exercise and Weight Loss Programs on Breast Cancer-Related Lymphedema Outcomes Among Over-weight Breast Cancer Survivors: The WISER Survivor Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1605–1613.
- Keith, L.; Rowsemitt, C.; Richards, L.G. Lifestyle Modification Group for Lymphedema and Obesity Results in Significant Health Outcomes. Am. J. Lifestyle Med. 2017, 14, 420–428.
- Liang, M.; Chen, Q.; Peng, K.; Deng, L.; He, L.; Hou, Y.; Zhang, Y.; Guo, J.; Mei, Z.; Li, L. Manual lymphatic drainage for lymphedema in patients after breast cancer surgery: A systematic review and meta-analysis of randomized controlled trials. Medicine 2020, 99, e23192.
- Muñoz-Alcaraz, M.N.; Pérula-De-Torres, L.; Serrano-Merino, J.; Jiménez-Vílchez, A.J.; Olmo-Carmona, M.V.; Muñoz-García, M.T.; Bartolomé-Moreno, C.; Oliván-Blázquez, B.; Magallón-Botaya, R. Efficacy and efficiency of a new therapeutic approach based on activity-oriented proprioceptive antiedema therapy (TAPA) for edema reduction and improved occupational performance in the rehabilitation of breast cancer-related arm lymphedema in women: A controlled, randomized clinical trial. BMC Cancer 2020, 20, 1–11.
- Barufi, S.; de Godoy, H.J.P.; de Godoy, J.M.P.; Godoy, M.D.F.G. Exercising and Compression Mechanism in the Treatment of Lymphedema. Cureus 2021, 13, e16121.
- Basta, M.; Gao, L.L.; Wu, L.C. Operative Treatment of Peripheral Lymphedema: A systematic meta-analysis of the efficacy and safety of lymphovenous microsurgery and tissue transplantation. Plast. Reconstr. Surg. 2014, 133, 905–913.
- Granzow, J.W.; Soderberg, J.M.; Kaji, A.H.; Dauphine, C. Review of Current Surgical Treatments for Lymphedema. Ann. Surg. Oncol. 2014, 21, 1195–1201.
- Raju, A.; Chang, D.W. Vascularized Lymph Node Transfer for Treatment of Lymphedema: A comprehensive literature review. Ann. Surg. 2015, 261, 1013–1023.
- Chang, E.I.; Schaverien, M.V.; Hanson, S.E.; Chu, C.K.; Hanasono, M.M. Evolution in Surgical Management of Breast Cancer-related Lymphedema: The MD Anderson Cancer Center Experience. Plast. Reconstr. Surg. Glob. Open 2020, 8, e2674.
- Hanson, S.E.; Chang, E.I.; Schaverien, M.V.; Chu, C.; Selber, J.C.; Hanasono, M.M. Controversies in Surgical Management of Lymphedema. Plast. Reconstr. Surg. Glob. Open 2020, 8, e2671.
- Gousopoulos, E.; Proulx, S.T.; Bachmann, S.B.; Scholl, J.; Dionyssiou, D.; Demiri, E.; Halin, C.; Dieterich, L.C.; Detmar, M. Regulatory T cell transfer ameliorates lymphedema and promotes lymphatic vessel function. JCI Insight 2016, 1, e89081.
- Gardenier, J.C.; Kataru, R.P.; Hespe, G.E.; Savetsky, I.; Torrisi, J.S.; Nores, G.D.G.; Jowhar, D.K.; Nitti, M.D.; Schofield, R.C.; Carlow, D.C.; et al. Topical tacrolimus for the treatment of secondary lymphedema. Nat. Commun. 2017, 8, 14345.
- Nores, G.D.G.; Ly, C.L.; Cuzzone, D.; Kataru, R.P.; Hespe, G.E.; Torrisi, J.S.; Huang, J.J.; Gardenier, J.C.; Savetsky, I.; Nitti, M.D.; et al. CD4+ T cells are activated in regional lymph nodes and migrate to skin to initiate lymphedema. Nat. Commun. 2018, 9, 1–14.
- Zampell, J.C.; Elhadad, S.; Avraham, T.; Weitman, E.; Aschen, S.; Yan, A.; Mehrara, B.J. Toll-like receptor deficiency worsens inflammation and lymphedema after lymphatic injury. Am. J. Physiol. Physiol. 2012, 302, C709–C719.
- Avraham, T.; Zampell, J.C.; Yan, A.; Elhadad, S.; Weitman, E.S.; Rockson, S.G.; Bromberg, J.; Mehrara, B.J. Th2 differentiation is necessary for soft tissue fibrosis and lymphatic dysfunction resulting from lymphedema. FASEB J. 2012, 27, 1114–1126.
- Mehrara, B.J.; Park, H.J.; Kataru, R.P.; Bromberg, J.; Coriddi, M.; Baik, J.E.; Shin, J.; Li, C.; Cavalli, M.R.; Encarnacion, E.M.; et al. Pilot Study of Anti-Th2 Immunotherapy for the Treatment of Breast Cancer-Related Upper Extremity Lymphedema. Biology 2021, 10, 934.
- Mazini, L.; Rochette, L.; Admou, B.; Amal, S.; Malka, G. Hopes and Limits of Adipose-Derived Stem Cells (ADSCs) and Mesenchymal Stem Cells (MSCs) in Wound Healing. Int. J. Mol. Sci. 2020, 21, 1306.
- Dave, M.; Mehta, K.; Luther, J.; Baruah, A.; Dietz, A.; Faubion, W.A. Mesenchymal Stem Cell Therapy for Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. Inflamm. Bowel Dis. 2015, 21, 2696–2707.
- Mishra, V.K.; Shih, H.-H.; Parveen, F.; Lenzen, D.; Ito, E.; Chan, T.-F.; Ke, L.-Y. Identifying the Therapeutic Significance of Mesenchymal Stem Cells. Cells 2020, 9, 1145.
- Rokunohe, A.; Matsuzaki, Y.; Rokunohe, D.; Sakuraba, Y.; Fukui, T.; Nakano, H.; Sawamura, D. Immunosuppressive effect of adipose-derived stromal cells on imiquimod-induced psoriasis in mice. J. Dermatol. Sci. 2015, 82, 50–53.
- Lee, Y.S.; Sah, S.K.; Lee, J.H.; Seo, K.-W.; Kang, K.-S.; Kim, T.-Y. Human umbilical cord blood-derived mesenchymal stem cells ameliorate psoriasis-like skin inflammation in mice. Biochem. Biophys. Rep. 2016, 9, 281–288.
- Jurado, M.; De La Mata, C.; Ruiz-García, A.; López-Fernández, E.; Espinosa, O.; Remigia, M.J.; Moratalla, L.; Goterris, R.; García-Martín, P.; Ruiz-Cabello, F.; et al. Adipose tissue-derived mesenchymal stromal cells as part of therapy for chronic graft-versus-host disease: A phase I/II study. Cytotherapy 2017, 19, 927–936.
- Conrad, C.; Niess, H.; Huss, R.; Huber, S.; Von Luettichau, I.; Nelson, P.J.; Ott, H.C.; Jauch, K.-W.; Bruns, C.J. Multipotent Mesenchymal Stem Cells Acquire a Lymphendothelial Phenotype and Enhance Lymphatic Regeneration In Vivo. Circulation 2009, 119, 281–289.
- Hwang, J.H.; Kim, I.G.; Lee, J.Y.; Piao, S.; Lee, D.S.; Lee, T.S.; Ra, J.C. Therapeutic lymphangiogenesis using stem cell and VEGF-C hydrogel. Biomaterials 2011, 32, 4415–4423.
- Zhou, H.; Wang, M.; Hou, C.; Jin, X.; Wu, X. Exogenous VEGF-C Augments the Efficacy of Therapeutic Lymphangiogenesis Induced by Allogenic Bone Marrow Stromal Cells in a Rabbit Model of Limb Secondary Lymphedema. Jpn. J. Clin. Oncol. 2011, 41, 841–846.
- Shimizu, Y.; Shibata, R.; Shintani, S.; Ishii, M.; Murohara, T. Therapeutic Lymphangiogenesis with Implantation of Adipose-Derived Regenerative Cells. J. Am. Heart Assoc. 2012, 1, e000877.
- Ackermann, M.; Wettstein, R.; Senaldi, C.; Kalbermatten, D.F.; Konerding, M.A.; Raffoul, W.; Erba, P. Impact of platelet rich plasma and adipose stem cells on lymphangiogenesis in a murine tail lymphedema model. Microvasc. Res. 2015, 102, 78–85.
- Yoshida, S.; Hamuy, R.; Hamada, Y.; Yoshimoto, H.; Hirano, A.; Akita, S. Adipose-derived stem cell transplantation for therapeutic lymphangiogenesis in a mouse secondary lymphedema model. Regen. Med. 2015, 10, 549–562.
- Hayashida, K.; Yoshida, S.; Yoshimoto, H.; Fujioka, M.; Saijo, H.; Migita, K.; Kumaya, M.; Akita, S. Adipose-Derived Stem Cells and Vascularized Lymph Node Transfers Successfully Treat Mouse Hindlimb Secondary Lymphedema by Early Reconnection of the Lymphatic System and Lymphangiogenesis. Plast. Reconstr. Surg. 2017, 139, 639–651.
- Beerens, M.; Aranguren, X.L.; Hendrickx, B.; Dheedene, W.; Dresselaers, T.; Himmelreich, U.; Verfaillie, C.; Luttun, A. Multipotent Adult Progenitor Cells Support Lymphatic Regeneration at Multiple Anatomical Levels during Wound Healing and Lymphedema. Sci. Rep. 2018, 8, 3852.
- Dai, T.; Jiang, Z.; Cui, C.; Sun, Y.; Lu, B.; Li, H.; Cao, W.; Chen, B.; Li, S.; Guo, L. The Roles of Podoplanin-Positive/Podoplanin-Negative Cells from Adipose-Derived Stem Cells in Lymphatic Regeneration. Plast. Reconstr. Surg. 2020, 145, 420–431.
- Ogino, R.; Hayashida, K.; Yamakawa, S.; Morita, E. Adipose-Derived Stem Cells Promote Intussusceptive Lymphangiogenesis by Restricting Dermal Fibrosis in Irradiated Tissue of Mice. Int. J. Mol. Sci. 2020, 21, 3885.
- Nguyen, D.; Zaitseva, T.S.; Zhou, A.; Rochlin, D.; Sue, G.; Deptula, P.; Tabada, P.; Wan, D.; Loening, A.; Paukshto, M.; et al. Lymphatic regeneration after implantation of aligned nanofibrillar collagen scaffolds: Preliminary preclinical and clinical results. J. Surg. Oncol. 2021, 125, 113–122.
- Scallan, J.P.; Zawieja, S.D.; Castorena-Gonzalez, J.A.; Davis, M.J. Lymphatic pumping: Mechanics, mechanisms and malfunction. J. Physiol. 2016, 594, 5749–5768.
- Chen, K.; Sinelnikov, M.Y.; Reshetov, I.V.; Timashev, P.; Gu, Y.; Mu, L.; Lu, P.; Zhang, Y. Therapeutic Potential of Mesenchymal Stem Cells for Postmastectomy Lymphedema: A Literature Review. Clin. Transl. Sci. 2020, 14, 54–61.
- Ly, C.L.; Nores, G.D.G.; Kataru, R.P.; Mehrara, B.J. T helper 2 differentiation is necessary for development of lymphedema. Transl. Res. 2018, 206, 57–70.
- Qiu, Y.; Chen, Y.; Fu, X.; Zhang, L.; Tian, J.; Hao, Q. HMGB1 promotes lymphangiogenesis of human lymphatic endothelial cells in vitro. Med. Oncol. 2010, 29, 358–363.
- Zampell, J.C.; Yan, A.; Avraham, T.; Andrade, V.; Malliaris, S.; Aschen, S.; Rockson, S.G.; Mehrara, B.J. Temporal and spatial patterns of endogenous danger signal expression after wound healing and in response to lymphedema. Am. J. Physiol. Physiol. 2011, 300, C1107–C1121.
- Ghanta, S.; Cuzzone, D.A.; Torrisi, J.S.; Albano, N.J.; Joseph, W.J.; Savetsky, I.L.; Gardenier, J.C.; Chang, D.; Zampell, J.C.; Mehrara, B.J. Regulation of inflammation and fibrosis by macrophages in lymphedema. Am. J. Physiol. Circ. Physiol. 2015, 308, H1065–H1077.
- Ogata, F.; Fujiu, K.; Matsumoto, S.; Nakayama, Y.; Shibata, M.; Oike, Y.; Koshima, I.; Watabe, T.; Nagai, R.; Manabe, I. Excess Lymphangiogenesis Cooperatively Induced by Macrophages and CD4+ T Cells Drives the Pathogenesis of Lymphedema. J. Investig. Dermatol. 2015, 136, 706–714.
- Gardenier, J.C.; Hespe, G.E.; Kataru, R.P.; Savetsky, I.; Torrisi, J.S.; Nores, G.D.G.; Dayan, J.J.; Chang, D.; Zampell, J.; Martinez-Corral, I.; et al. Diphtheria toxin–mediated ablation of lymphatic endothelial cells results in progressive lymphedema. JCI Insight 2016, 1, e84095.
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362.
- Tal, O.; Lim, H.Y.; Gurevich, I.; Milo, I.; Shipony, Z.; Ng, L.G.; Angeli, V.; Shakhar, G. DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J. Exp. Med. 2011, 208, 2141–2153.
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; de Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial Dendritic Cell Guidance by Haptotactic Chemokine Gradients. Science 2013, 339, 328–332.
- Teijeira, A.; Rouzaut, A.; Melero, I. Initial Afferent Lymphatic Vessels Controlling Outbound Leukocyte Traffic from Skin to Lymph Nodes. Front. Immunol. 2013, 4, 433.
- Russo, E.; Teijeira, A.; Vaahtomeri, K.; Willrodt, A.-H.; Bloch, J.; Nitschké, M.; Santambrogio, L.; Kerjaschki, D.; Sixt, M.; Halin, C. Intralymphatic CCL21 Promotes Tissue Egress of Dendritic Cells through Afferent Lymphatic Vessels. Cell Rep. 2016, 14, 1723–1734.
- Syrbe, U.; Siveke, J.; Hamann, A. Th1/Th2 subsets: Distinct differences in homing and chemokine receptor expression? Springer Semin. Immunopathol. 1999, 21, 263–285.
- Reiss, Y.; Proudfoot, A.E.; Power, C.A.; Campbell, J.; Butcher, E.C. CC Chemokine Receptor (CCR)4 and the CCR10 Ligand Cutaneous T Cell–attracting Chemokine (CTACK) in Lymphocyte Trafficking to Inflamed Skin. J. Exp. Med. 2001, 194, 1541–1547.
- Agace, W.W. Tissue-tropic effector T cells: Generation and targeting opportunities. Nat. Rev. Immunol. 2006, 6, 682–692.
- Ni, Z.; Walcheck, B. Cutaneous lymphocyte-associated antigen (CLA) T cells up-regulate P-selectin ligand expression upon their activation. Clin. Immunol. 2009, 133, 257–264.
- Ly, C.; Cuzzone, D.; Kataru, R.P.; Mehrara, B.J. Small Numbers of CD4+ T Cells Can Induce Development of Lymphedema. Plast. Reconstr. Surg. 2019, 143, 518e–526e.
- Chiaramonte, M.G.; Cheever, A.W.; Malley, J.D.; Donaldson, D.D.; Wynn, T.A. Studies of murine schistosomiasis reveal interleukin-13 blockade as a treatment for established and progressive liver fibrosis. Hepatology 2001, 34, 273–282.
- Wynn, T.A. Fibrotic disease and the TH1/TH2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594.
- Clavin, N.W.; Avraham, T.; Fernandez, J.; Daluvoy, S.V.; Soares, M.A.; Chaudhry, A.; Mehrara, B.J. TGF-β1 is a negative regulator of lymphatic regeneration during wound repair. Am. J. Physiol. Circ. Physiol. 2008, 295, H2113–H2127.
- Kataru, R.P.; Kim, H.; Jang, C.; Choi, D.K.; Koh, B.I.; Kim, M.; Gollamudi, S.; Kim, Y.-K.; Lee, S.-H.; Koh, G.Y. T Lymphocytes Negatively Regulate Lymph Node Lymphatic Vessel Formation. Immunity 2011, 34, 96–107.
- Kim, H.; Kataru, R.; Koh, G.Y. Inflammation-associated lymphangiogenesis: A double-edged sword? J. Clin. Investig. 2014, 124, 936–942.
- Shin, K.; Kataru, R.P.; Park, H.J.; Kwon, B.-I.; Kim, T.W.; Hong, Y.K.; Lee, S.-H. TH2 cells and their cytokines regulate formation and function of lymphatic vessels. Nat. Commun. 2015, 6, 6196.
- Savetsky, I.L.; Ghanta, S.; Gardenier, J.C.; Torrisi, J.S.; Nores, G.D.G.; Hespe, G.E.; Nitti, M.D.; Kataru, R.P.; Mehrara, B.J. Th2 Cytokines Inhibit Lymphangiogenesis. PLoS ONE 2015, 10, e0126908.
- Karlsen, T.V.; Karkkainen, M.J.; Alitalo, K.; Wiig, H. Transcapillary fluid balance consequences of missing initial lymphatics studied in a mouse model of primary lymphoedema. J. Physiol. 2006, 574, 583–596.
- Cuzzone, D.A.; Weitman, E.S.; Albano, N.J.; Ghanta, S.; Savetsky, I.; Gardenier, J.C.; Joseph, W.J.; Torrisi, J.S.; Bromberg, J.F.; Olszewski, W.; et al. IL-6 regulates adipose deposition and homeostasis in lymphedema. Am. J. Physiol. Circ. Physiol. 2014, 306, H1426–H1434.
- Torrisi, J.S.; Hespe, G.E.; Cuzzone, D.A.; Savetsky, I.L.; Nitti, M.D.; Gardenier, J.C.; Nores, G.D.G.; Jowhar, D.; Kataru, R.; Mehrara, B.J. Inhibition of Inflammation and iNOS Improves Lymphatic Function in Obesity. Sci. Rep. 2016, 6, 19817.
- Mand, S.; Debrah, A.; Klarmann, U.; Batsa, L.; Marfo-Debrekyei, Y.; Kwarteng, A.; Specht, S.; Belda-Domene, A.; Fimmers, R.; Taylor, M.; et al. Doxycycline Improves Filarial Lymphedema Independent of Active Filarial Infection: A Randomized Controlled Trial. Clin. Infect. Dis. 2012, 55, 621–630.
- Henehan, M.; Montuno, M.; De Benedetto, A. Doxycycline as an anti-inflammatory agent: Updates in dermatology. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1800–1808.
- Nakamura, K.; Radhakrishnan, K.; Wong, Y.M.; Rockson, S.G. Anti-Inflammatory Pharmacotherapy with Ketoprofen Ameliorates Experimental Lymphatic Vascular Insufficiency in Mice. PLoS ONE 2009, 4, e8380.
- Rockson, S.G.; Tian, W.; Jiang, X.; Kuznetsova, T.; Haddad, F.; Zampell, J.; Mehrara, B.; Sampson, J.P.; Roche, L.; Kim, J.; et al. Pilot studies demonstrate the potential benefits of antiinflammatory therapy in human lymphedema. JCI Insight 2018, 3, e123775.
- Tian, W.; Rockson, S.G.; Jiang, X.; Kim, J.; Begaye, A.; Shuffle, E.M.; Tu, A.B.; Cribb, M.; Nepiyushchikh, Z.; Feroze, A.H.; et al. Leukotriene B4 antagonism ameliorates experimental lymphedema. Sci. Transl. Med. 2017, 9, eaal3920.
- Cribb, M.; Sestito, L.; Rockson, S.; Nicolls, M.; Thomas, S.; Dixon, J. The Kinetics of Lymphatic Dysfunction and Leukocyte Expansion in the Draining Lymph Node during LTB4 Antagonism in a Mouse Model of Lymphedema. Int. J. Mol. Sci. 2021, 22, 4455.
- Kasseroller, R.G.; Schrauzer, G.N. Treatment of Secondary Lymphedema of the Arm with Physical Decongestive Therapy and Sodium Selenite: A review. Am. J. Ther. 2000, 7, 273–279.
- Bruns, F.; Micke, O.; Bremer, M. Current status of selenium and other treatments for secondary lymphedema. J. Support. Oncol. 2004, 1, 121–130.
- Han, H.W.; Yang, E.J.; Lee, S.-M. Sodium Selenite Alleviates Breast Cancer-Related Lymphedema Independent of Antioxidant Defense System. Nutrients 2019, 11, 1021.
- Lee, H.; Lee, B.; Kim, Y.; Min, S.; Yang, E.; Lee, S. Effects of Sodium Selenite Injection on Serum Metabolic Profiles in Women Diagnosed with Breast Cancer-Related Lymphedema—Secondary Analysis of a Randomized Placebo-Controlled Trial Using Global Metabolomics. Nutrients 2021, 13, 3253.
- McLoughlin, R.F.; Berthon, B.S.; Jensen, M.E.; Baines, K.J.; Wood, L.G. Short-chain fatty acids, prebiotics, synbiotics, and systemic inflammation: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2017, 106, 930–945.
- Vafa, S.; Haghighat, S.; Janani, L.; Totmaj, A.S.; Navaei, M.; Amirinejad, A.; Emamat, H.; Salehi, Z.; Zarrati, M. The effects of synbiotic supplementation on serum inflammatory markers and edema volume in breast cancer survivors with lymphedema. EXCLI J. 2020, 19, 1–15.
- Vafa, S.; Zarrati, M.; Malakootinejad, M.; Totmaj, A.S.; Zayeri, F.; Salehi, M.; Sanati, V.; Haghighat, S. Calorie restriction and synbiotics effect on quality of life and edema reduction in breast cancer-related lymphedema, a clinical trial. Breast 2020, 54, 37–45.
- Navaei, M.; Haghighat, S.; Janani, L.; Vafa, S.; Totmaj, A.S.; Lahiji, M.R.; Emamat, H.; Salehi, Z.; Amirinejad, A.; Izad, M.; et al. The Effects of Synbiotic Supplementation on Antioxidant Capacity and Arm Volumes in Survivors of Breast Cancer-Related Lymphedema. Nutr. Cancer 2019, 72, 62–73.
- Jorgensen, K.A.; Koefoed-Nielsen, P.B.; Karamperis, N. Calcineurin Phosphatase Activity and Immunosuppression. A Review on the Role of Calcineurin Phosphatase Activity and the Immunosuppressive Effect of Cyclosporin A and Tacrolimus. Scand. J. Immunol. 2003, 57, 93–98.
- McKinstry, K.K.; Strutt, T.M.; Bautista, B.; Zhang, W.; Kuang, Y.; Cooper, A.; Swain, S.L. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat. Commun. 2014, 5, 5377.
- Yoon, S.-H.; Kim, K.Y.; Wang, Z.; Park, J.-H.; Bae, S.M.; Kim, S.-Y.; Song, H.-Y.; Jeon, J.Y. EW-7197, a Transforming Growth Factor-Beta Type I Receptor Kinase Inhibitor, Ameliorates Acquired Lymphedema in a Mouse Tail Model. Lymphat. Res. Biol. 2020, 18, 433–438.
- Avraham, T.; Yan, A.; Zampell, J.C.; Daluvoy, S.V.; Haimovitz-Friedman, A.; Cordeiro, A.P.; Mehrara, B.J. Radiation therapy causes loss of dermal lymphatic vessels and interferes with lymphatic function by TGF-β1-mediated tissue fibrosis. Am. J. Physiol. Physiol. 2010, 299, C589–C605.
This entry is offline, you can click here to edit this entry!