The presence of the IDH mutation in glioma raises the possibility that these CNS malignancies could be targeted with metabolic-based therapeutics. Exploration of the role that regulatory lipids, such as sphingolipids serve within the IDH1mut gliomas is limited. Our study incorporates LC/MS lipidomic discovery with an aim to identify vulnerabilities within the sphingolipid metabolism that could be exploited therapeutically. We reveal elevation in certain lipids produced along the sphingolipid degradation pathway for IDH1 mutated glioma cells. Our review of TCGA data from clinical patients reveal a parallel trend for enzymes associated with sphingolipid degradation that correlates to increased survivability in glioma patients. We demonstrate the biostatic effects of treatments which target this pathway via inhibiting sphingosine kinase and exacerbating the imbalance between sphingosine and sphingosine 1-phosphate.
- S1P
- IDH1mut
- astrocytoma
- oligodendroglioma
- Sphingosine
- ceramide
- N,N-dimethylsphingosine
- sphingosine kinase inhibitor
- sphingolipid-rheostat balance
- lipidomic
1. Introduction
Gliomas are devastating brain tumors with no known curable treatment. Despite decades of progress in this area, the overall survival prognoses for patients diagnosed with these tumors have not improved drastically. Gain-of-function mutations in isocitrate dehydrogenase I (IDHmut) are very common events in lower grade gliomas and secondary glioblastomas, causing overproduction of D-2-hydroxyglutarate at millimolar concentrations [1,2,3]. Although, overall patients with low-grade IDHmut gliomas are known to present increased survival compared to both low- and high-grade glioblastomas, this survival could be improved with additional molecular therapies [4,5,6]. The presence of a metabolic alteration in these gliomas raises the possibility of implementing metabolic-based therapeutics; however, a comprehensive understanding of metabolic vulnerabilities within the IDH1mut gliomas is limited. Since IDHWT is a central enzyme of the tricarboxylic acid (TCA) cycle, many investigations have explored targeting of central carbon metabolism in these tumors. As a result, several targeting approaches have been proposed within related pathways which include targeting glutaminase [2,7], nicotinamide adenine dinucleotide (NAD) metabolism [8], exploiting the increased reactive oxygen species (ROS) intrinsically present in IDH1mut gliomas [9,10], as well as the direct inhibition of IDH1R132H mutant activity [10]. While the central carbon metabolism has been studied extensively in cancer, and specifically in gliomas, the lipidomic analysis is still sparse. Very few investigations are focused on lipids while even fewer are focused on sphingolipids within the IDH1mut gliomas, despite their important roles in oncogenic signaling [11].
Ceramide and sphingosine 1-phosphate (S1P) are amongst the most studied sphingolipids. Ceramide and sphingosine maintain a negative correlation with S1P via the sphingolipid-rheostat balance, in which the upregulation of ceramide is directly correlated to a concomitant downregulation of S1P and vice versa [12]. The fate and accumulation of sphingosine is a major driver in the sphingolipid rheostat balance, which directly modulates cell fate [11,13]. Moreover, sphingosine has been implicated in anti-proliferative signaling in a manner that resembles ceramide-induced apoptosis in certain cancers [12,14]. Sphingosine can inhibit protein kinase C and phosphatidic acid phosphohydrolase, whose activities promote the oncoprotective, anti-apoptotic response in certain cancers [15,16]. Moreover, sphingosine stimulates phospholipase C-protein kinase C-mediated activation of phospholipase D, which is attributed to inducing apoptosis in certain cancer cells [17].
While certain sphingolipids (e.g., analogs of ceramide, sphingosine and sphinganine) can serve as bioeffectors that disrupt pro-survival cellular signaling pathways, S1P has been classified as an oncopotent biomolecule which mediates cell signaling as an intracellular second messenger and a ligand of G protein-coupled receptors (GPCR) [18,19]. S1P utilizes multiple targets to stimulate complex cellular processes involving chemotactic motility, migration, differentiation, proliferation, apoptotic survival response, capillary permeability, differentiation and angiogenesis, which are key hallmarks of cancers [18]. Noteworthy, endogenous analogs of S1P (phytosphingosine 1-phosphate, dihydrosphingosine 1-phosphate and ceramide 1-phosphate) are also bioeffectors that modulate the pro-survival response in tumors, especially [19,20,21]. Despite known roles of ceramide, sphingosine and S1P in multiple cancer types, their contribution to IDH1mut gliomas remains unknown. Given their historical, vital roles in cell signaling and regulation, we hypothesized that manipulating the dynamic relationship between ceramide, sphingosine and S1P would reveal vulnerabilities and potentially translate into therapeutic targets to combat proliferation in IDH1mut gliomas.
2. Targeting the Sphingolipid Pathway in IDH1mut Glioma Cells
Having demonstrated that the IDH1mut glioma cell sphingolipid profile contains high sphingosine/ceramide and low S1P from both metabolite as well as enzyme overexpression analyses, we next targeted the sphingolipid pathway with the goal to determine a combination of compounds that effectively arrests tumor cell proliferation and leads to cell death. We initially tested an inhibitor of sphingosine kinase I (SphK1), PF-543, to demonstrate the feasibility of enhancing this potential metabolic vulnerability by further decreasing intracellular levels of S1P. Morphologically, the BT142 cell line displayed a higher sensitivity to PF-543 as seen in the optical images (Figure S2). Given the promising results of inhibiting SphK1 in two IDH1mut cell lines (BT142 and TS603), we expanded the panel to include additional SphK inhibitors (SpKi) and other sphingolipid-based bioeffectors. Therefore, we designed a small scale, rational drug screen of 17 related compounds including commercially available SphK inhibitors as well as specific endogenous bioeffectors, such as NDMS, sphingosine-C17 (Sph C17) and sphinganine-C18 (dhSph C18), which were determined to be elevated in our metabolic analysis of IDH1mut neurospheres. By treating IDH1mut cells with these sphingoid bioeffectors, we rationalized that this approach would further enhance the metabolic/cellular stress already existent due to their intrinsic elevation in IDHmut glioma lines. In the rational drug panel, we also included ceramide C16, which was previously reported to promote growth [19] as well as cisplatin, which was reported to activate acid sphingomyelinase (SMPD2) [26]. Amongst the 17 compounds, NDMS, sphingosine-C17 and sphinganine-C18 were consistently effective and potent in both representative IDH1mut glioma cell lines (Figure 4a,b). Interestingly, NDMS, which is an endogenous bioeffector that inhibits SphK, elicited a potent biostatic effect that exceeded most of the synthetic SphK inhibitors present in our rational panel, particularly PF543 used in the pilot experiment.
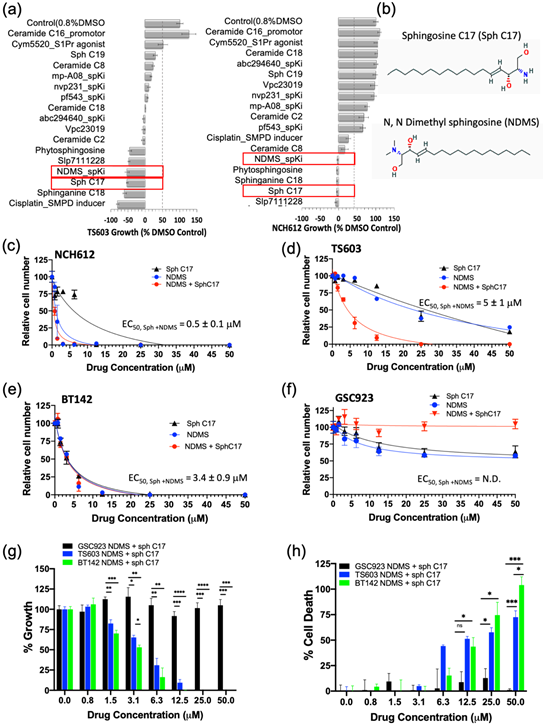
Since the elevation of endogenous bioeffectors, NDMS and sphingosine C17 elicited a highly potent biostatic response in two IDH1mut cell lines, we decided to determine their dose-dependent effects on cell viability (proliferation) and cytotoxicity (cell death). Both the cell proliferation and cell death assays confirmed that all of the tested IDH1mut cells presented a dose-dependent response to treatment with the NDMS and sphingosine and produced a greater effect when combined. In particular, the oligodendroglioma cell lines (TS603 and NCH612) displayed an increased effect by combining both NDMS and Sph C17 while the astrocytoma cell line (BT142) was equally sensitive to independent versus combined treatment (Figure 4c–e, Table S5). By contrast, the IDH1WT cell line (GSC923) presented little sensitivity to these drug or combination treatments (Figure 4f–h, Table S5).
The response to the combinatory treatment suggests high specificity to IDH1mut cell lines both on the growth of the cells (Figure 4g) as well as the percentage of cellular death (Figure 4h). Collectively, these findings suggest a model in which targeting sphingolipid metabolism by elevating sphingosine and inhibiting SPHK with N,N-dimethylsphingosine presents a remarkable vulnerability of IDH1mut gliomas. Further investigation of the downstream sphingolipid signaling events that are disrupted in tandem with this treatment is expected to reveal additional vulnerable targets in the cell-regulation of IDH1mut gliomas.
Reference
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y. Clinical impact of revisions to the WHO classification of diffuse gliomas and associated future problems. Int. J. Clin. Oncol. 2020, 25, 1004–1009. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e125. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- Mora, R.; Dokic, I.; Kees, T.; Hüber, C.M.; Keitel, D.; Geibig, R.; Brügge, B.; Zentgraf, H.; Brady, N.R.; Régnier-Vigouroux, A. Sphingolipid rheostat alterations related to transformation can be exploited for specific induction of lysosomal cell death in murine and human glioma. Glia 2010, 58, 1364–1383. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.R.; Williams, J.B. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: Oxidative stress and the seesaw of cell survival and death. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2012, 163, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef]
- Lai, M.; La Rocca, V.; Amato, R.; Freer, G.; Pistello, M. Sphingolipid/Ceramide Pathways and Autophagy in the Onset and Progression of Melanoma: Novel Therapeutic Targets and Opportunities. Int. J. Mol. Sci. 2019, 20, 3436. [Google Scholar] [CrossRef] [PubMed]
- Mullmann, T.J.; Siegel, M.I.; Egan, R.W.; Billah, M.M. Sphingosine inhibits phosphatidate phosphohydrolase in human neutrophils by a protein kinase C-independent mechanism. J. Biol. Chem. 1991, 266, 2013–2016. [Google Scholar] [PubMed]
- Meana, C.; García-Rostán, G.; Peña, L.; Lordén, G.; Cubero, Á.; Orduña, A.; Győrffy, B.; Balsinde, J.; Balboa, M.A. The phosphatidic acid phosphatase lipin-1 facilitates inflammation-driven colon carcinogenesis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.H.; Kim, S.Y.; Kim, E.H.; Choi, K.S.; Kwon, T.K.; Lee, Y.H.; Chang, J.S.; Kim, M.S.; Jo, Y.H.; Min, D.S. Transmodulation between phospholipase D and c-Src enhances cell proliferation. Mol. Cell. Biol. 2003, 23, 3103–3115. [Google Scholar] [CrossRef] [PubMed]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010, 6, 1603–1624. [Google Scholar] [CrossRef]
- Berdyshev, E.V.; Gorshkova, I.A.; Usatyuk, P.; Zhao, Y.; Saatian, B.; Hubbard, W.; Natarajan, V. De novo biosynthesis of dihydrosphingosine-1-phosphate by sphingosine kinase 1 in mammalian cells. Cell. Signal. 2006, 18, 1779–1792. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Lita, A.; Pliss, A.; Kuzmin, A.; Yamasaki, T.; Zhang, L.; Dowdy, T.; Burks, C.; de Val, N.; Celiku, O.; Ruiz-Rodado, V.; et al. IDH1 Mutations Induce Organelle Defects Via Dysregulated Phospholipids. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef] [PubMed]
- Jabalee, J.; Towle, R.; Lawson, J.; Dickman, C.; Garnis, C. Sphingomyelin phosphodiesterase 3 methylation and silencing in oral squamous cell carcinoma results in increased migration and invasion and altered stress response. Oncotarget 2020, 11, 523–534. [Google Scholar] [CrossRef]
- Lacour, S.; Hammann, A.; Grazide, S.; Lagadic-Gossmann, D.; Athias, A.; Sergent, O.; Laurent, G.; Gambert, P.; Solary, E.; Dimanche-Boitrel, M.T. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 2004, 64, 3593–3598. [Google Scholar] [CrossRef] [PubMed]
This entry is adapted from the peer-reviewed paper 10.3390/cancers12102910