Named as the guardian of the genome, p53 is a tumor suppressor that regulates cell function, often through many different mechanisms such as DNA repair, apoptosis, cell cycle arrest, senescence, metabolism, and autophagy. One of the genes that p53 activates is MDM2, which forms a negative feedback loop since MDM2 induces the degradation of p53. When p53 activity is inhibited, damaged cells do not undergo cell cycle arrest or apoptosis. As 50% of human cancers inactivate p53 by mutation, current research focuses on reactivating p53 by developing drugs that target the p53-MDM2 interaction, which includes the binding of MDM2 and phosphorylation of p53.
- p53
- Mdm2
- cancer
- small molecules
- chemotherapy
1. Introduction
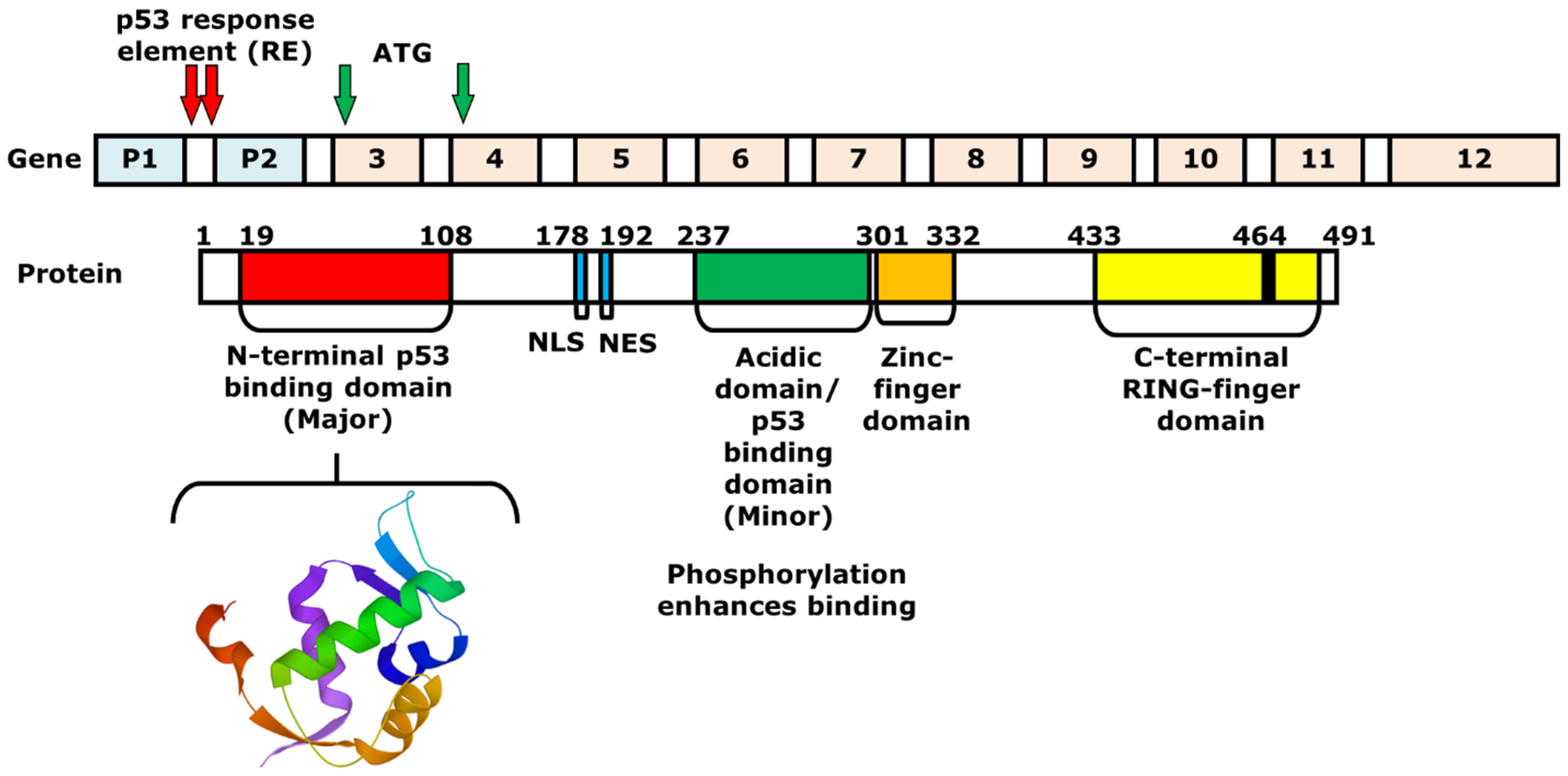
2. Mechanism of p53 and MDM2-Binding
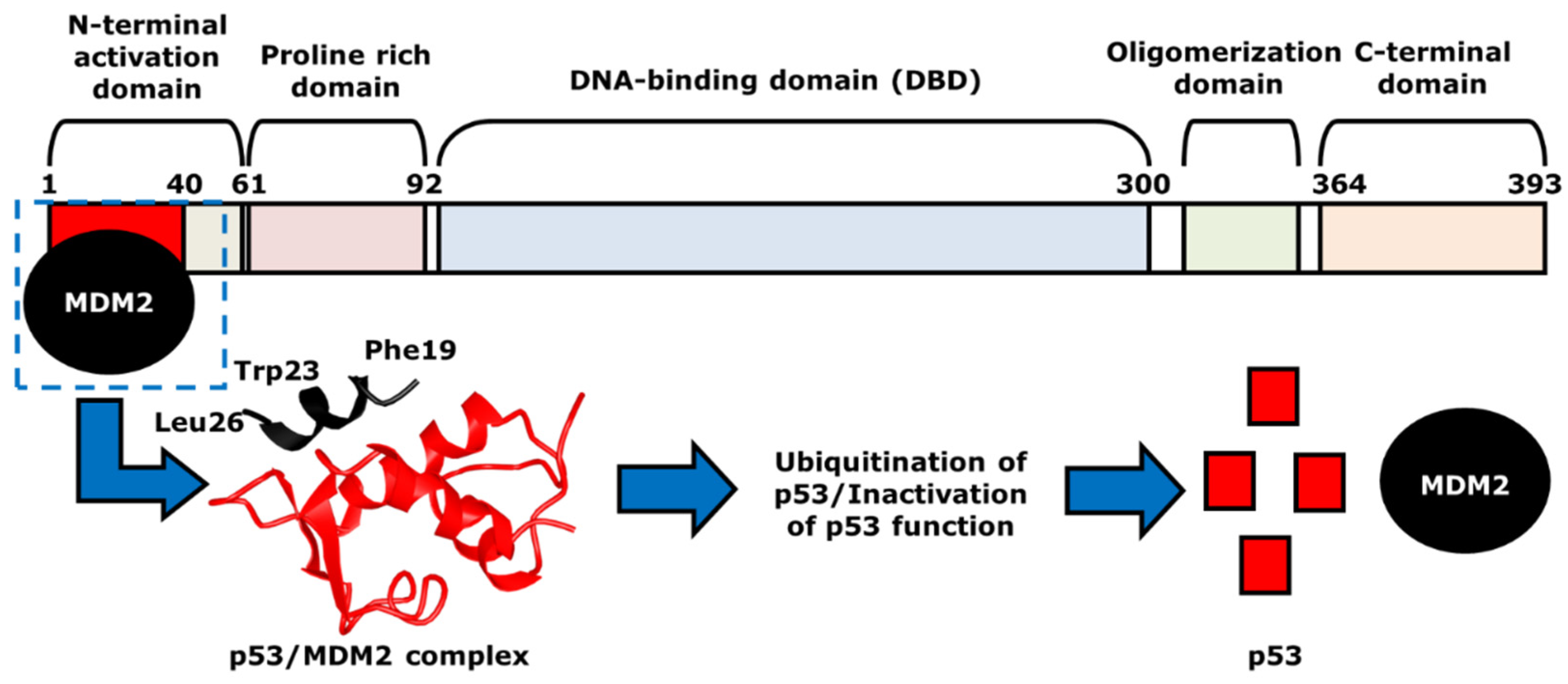
3. The Role of p53 in Cancer Chemotherapy
4. Strategies of Blocking the p53/MDM2 Interaction
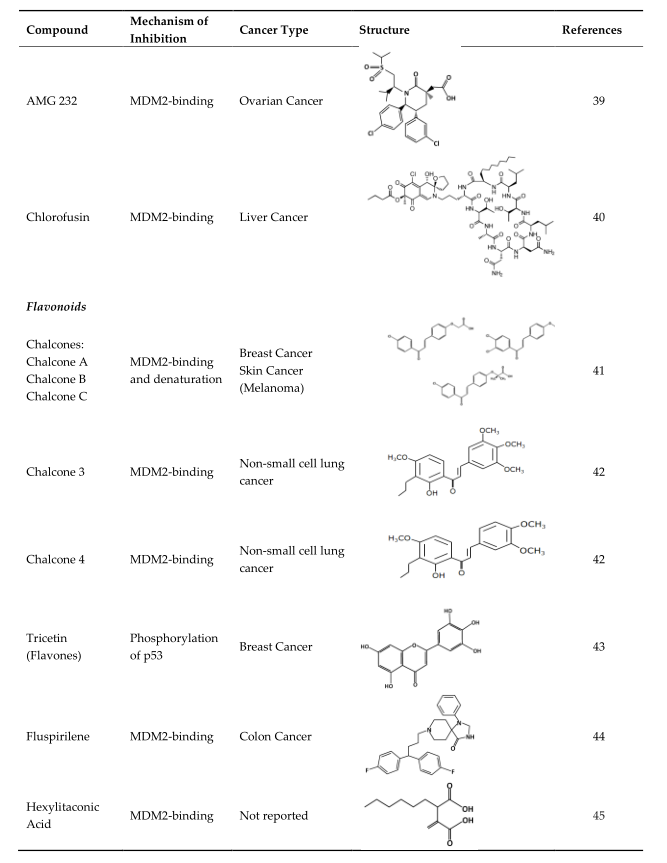
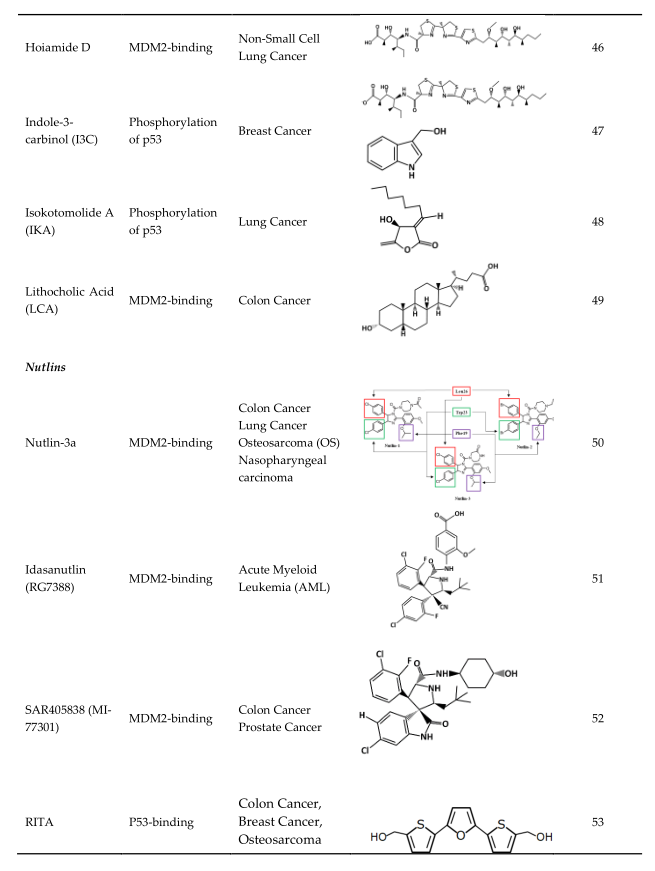
This entry is adapted from the peer-reviewed paper 10.3390/ijms23095005
References
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nature reviews Cancer. 2014;14(5):359-70. doi: 10.1038/nrc3711. PubMed PMID: 24739573; PubMed Central PMCID: PMC4049238.
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nature reviews Cancer. 2002;2(8):594-604. doi: 10.1038/nrc864. PubMed PMID: 12154352.
- Chen J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb Perspect Med. 2016;6(3):a026104. doi: 10.1101/cshperspect.a026104. PubMed PMID: 26931810; PubMed Central PMCID: PMCPMC4772082.
- Vousden KH, Ryan KM. p53 and metabolism. Nature reviews Cancer. 2009;9(10):691-700. doi: 10.1038/nrc2715. PubMed PMID: 19759539.
- White E. Autophagy and p53. Cold Spring Harb Perspect Med. 2016;6(4):a026120. doi: 10.1101/cshperspect.a026120. PubMed PMID: 27037419; PubMed Central PMCID: PMCPMC4817743.
- Guo JY, White E. Autophagy, Metabolism, and Cancer. Cold Spring Harbor symposia on quantitative biology. 2016;81:73-8. doi: 10.1101/sqb.2016.81.030981. PubMed PMID: 28209717; PubMed Central PMCID: PMCPMC5521269.
- Fernandez-Fernandez MR, Sot B. The relevance of protein-protein interactions for p53 function: the CPE contribution. Protein engineering, design & selection : PEDS. 2011;24(1-2):41-51. doi: 10.1093/protein/gzq074. PubMed PMID: 20952436.
- Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Molecular and cellular biology. 2007;27(3):1056-68. doi: 10.1128/MCB.01307-06. PubMed PMID: 17116689; PubMed Central PMCID: PMC1800693.
- Iwakuma T, Lozano G. MDM2, an introduction. Molecular cancer research : MCR. 2003;1(14):993-1000. PubMed PMID: 14707282.
- Loh SN. The missing zinc: p53 misfolding and cancer. Metallomics : integrated biometal science. 2010;2(7):442-9. doi: 10.1039/c003915b. PubMed PMID: 21072344.
- Perry ME, Mendrysa SM, Saucedo LJ, Tannous P, Holubar M. p76(MDM2) inhibits the ability of p90(MDM2) to destabilize p53. The Journal of biological chemistry. 2000;275(8):5733-8. doi: 10.1074/jbc.275.8.5733. PubMed PMID: 10681559.
- Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123(1):49-63. doi: 10.1016/j.cell.2005.07.034. PubMed PMID: 16213212.
- Manfredi JJ. Mdm2 and MdmX: Partners in p53 Destruction. Cancer research. 2021;81(7):1633-4. doi: 10.1158/0008-5472.CAN-21-0145. PubMed PMID: 34003788.
- Moll UM, Petrenko O. The MDM2-p53 interaction. Molecular cancer research : MCR. 2003;1(14):1001-8. PubMed PMID: 14707283.
- Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annual review of pharmacology and toxicology. 1999;39:295-312. doi: 10.1146/annurev.pharmtox.39.1.295. PubMed PMID: 10331086.
- Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutation research. 2010;704(1-3):12-20. doi: 10.1016/j.mrrev.2010.01.009. PubMed PMID: 20096807.
- Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor perspectives in biology. 2013;5(9). doi: 10.1101/cshperspect.a012716. PubMed PMID: 24003211; PubMed Central PMCID: PMCPMC3753707.
- Lanz MC, Dibitetto D, Smolka MB. DNA damage kinase signaling: checkpoint and repair at 30 years. The EMBO journal. 2019;38(18):e101801. doi: 10.15252/embj.2019101801. PubMed PMID: 31393028; PubMed Central PMCID: PMCPMC6745504.
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in cancer research. 2010;108:73-112. doi: 10.1016/B978-0-12-380888-2.00003-0. PubMed PMID: 21034966.
- Ou YH, Chung PH, Sun TP, Shieh SY. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Molecular biology of the cell. 2005;16(4):1684-95. doi: 10.1091/mbc.e04-08-0689. PubMed PMID: 15659650; PubMed Central PMCID: PMC1073652.
- Marei HE, Althani A, Afifi N, Hasan A, Caceci T, Pozzoli G, et al. p53 signaling in cancer progression and therapy. Cancer cell international. 2021;21(1):703. doi: 10.1186/s12935-021-02396-8. PubMed PMID: 34952583; PubMed Central PMCID: PMCPMC8709944.
- Hill R, Bodzak E, Blough MD, Lee PW. p53 Binding to the p21 promoter is dependent on the nature of DNA damage. Cell cycle. 2008;7(16):2535-43. doi: 10.4161/cc.7.16.6440. PubMed PMID: 18719376.
- Kachnic LA, Wu B, Wunsch H, Mekeel KL, DeFrank JS, Tang W, et al. The ability of p53 to activate downstream genes p21(WAF1/cip1) and MDM2, and cell cycle arrest following DNA damage is delayed and attenuated in scid cells deficient in the DNA-dependent protein kinase. The Journal of biological chemistry. 1999;274(19):13111-7. doi: 10.1074/jbc.274.19.13111. PubMed PMID: 10224064.
- Gartel AL. Is p21 an oncogene? Molecular cancer therapeutics. 2006;5(6):1385-6. doi: 10.1158/1535-7163.MCT-06-0163. PubMed PMID: 16818495.
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife. 2014;3. doi: 10.7554/eLife.02872. PubMed PMID: 24876129; PubMed Central PMCID: PMC4076869.
- Day PJ, Cleasby A, Tickle IJ, O'Reilly M, Coyle JE, Holding FP, et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(11):4166-70. doi: 10.1073/pnas.0809645106. PubMed PMID: 19237565; PubMed Central PMCID: PMC2657441.
- Surova O, Zhivotovsky B. Various modes of cell death induced by DNA damage. Oncogene. 2013;32(33):3789-97. doi: 10.1038/onc.2012.556. PubMed PMID: 23208502.
- De Zio D, Cianfanelli V, Cecconi F. New insights into the link between DNA damage and apoptosis. Antioxidants & redox signaling. 2013;19(6):559-71. doi: 10.1089/ars.2012.4938. PubMed PMID: 23025416; PubMed Central PMCID: PMCPMC3717195.
- Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annual review of pharmacology and toxicology. 2009;49:223-41. doi: 10.1146/annurev.pharmtox.48.113006.094723. PubMed PMID: 18834305; PubMed Central PMCID: PMC2676449.
- Wang S, Zhao Y, Aguilar A, Bernard D, Yang CY. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb Perspect Med. 2017;7(5). doi: 10.1101/cshperspect.a026245. PubMed PMID: 28270530; PubMed Central PMCID: PMCPMC5411684.
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281(5383):1674-7. doi: 10.1126/science.281.5383.1674. PubMed PMID: 9733514.
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91(3):325-34. doi: 10.1016/s0092-8674(00)80416-x. PubMed PMID: 9363941.
- Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes & development. 2000;14(3):278-88. PubMed PMID: 10673500; PubMed Central PMCID: PMCPMC316357.
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes & development. 2000;14(3):289-300. PubMed PMID: 10673501; PubMed Central PMCID: PMCPMC316358.
- Chen J. The Roles of MDM2 and MDMX Phosphorylation in Stress Signaling to p53. Genes Cancer. 2012;3(3-4):274-82. doi: 10.1177/1947601912454733. PubMed PMID: 23150760; PubMed Central PMCID: PMCPMC3494364.
- Freedman DA, Epstein CB, Roth JC, Levine AJ. A genetic approach to mapping the p53 binding site in the MDM2 protein. Molecular medicine. 1997;3(4):248-59. PubMed PMID: 9131587; PubMed Central PMCID: PMCPMC2230064.
- Beloglazkina A, Zyk N, Majouga A, Beloglazkina E. Recent Small-Molecule Inhibitors of the p53-MDM2 Protein-Protein Interaction. Molecules. 2020;25(5). doi: 10.3390/molecules25051211. PubMed PMID: 32156064; PubMed Central PMCID: PMCPMC7179467.
- Sahin I, Zhang S, Navaraj A, Zhou L, Dizon D, Safran H, et al. AMG-232 sensitizes high MDM2-expressing tumor cells to T-cell-mediated killing. Cell Death Discov. 2020;6:57. doi: 10.1038/s41420-020-0292-1. PubMed PMID: 32655895; PubMed Central PMCID: PMC7338458.
- Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. Journal of medicinal chemistry. 2014;57(4):1454-72. doi: 10.1021/jm401753e. PubMed PMID: 24456472.
- Duncan SJ, Gruschow S, Williams DH, McNicholas C, Purewal R, Hajek M, et al. Isolation and structure elucidation of Chlorofusin, a novel p53-MDM2 antagonist from a Fusarium sp. Journal of the American Chemical Society. 2001;123(4):554-60. doi: 10.1021/ja002940p. PubMed PMID: 11456567.
- Balentine DA, Dwyer JT, Erdman JW, Jr., Ferruzzi MG, Gaine PC, Harnly JM, et al. Recommendations on reporting requirements for flavonoids in research. Am J Clin Nutr. 2015;101(6):1113-25. doi: 10.3945/ajcn.113.071274. PubMed PMID: 25854881.
- Pereira D LR, Palmeira A, Seca H, Soares J, Gomes S, Raimundo L, Maciel C, Pinto M, Sousa E, Vasconcelos MH, Saraiva L, Cidade H. Design and synthesis of new inhibitors of p53–MDM2 interaction with a chalcone scaffold. Arabian Journal of Chemistry. 2019;12(8):12. doi: doi.org/10.1016/j.arabjc.2016.04.015.
- Qin JJ, Li X, Hunt C, Wang W, Wang H, Zhang R. Natural products targeting the p53-MDM2 pathway and mutant p53: Recent advances and implications in cancer medicine. Genes & diseases. 2018;5(3):204-19. doi: 10.1016/j.gendis.2018.07.002. PubMed PMID: 30320185; PubMed Central PMCID: PMC6176154.
- Tichy A, Vavrova J, Pejchal J, Rezacova M. Ataxia-telangiectasia mutated kinase (ATM) as a central regulator of radiation-induced DNA damage response. Acta medica. 2010;53(1):13-7. doi: 10.14712/18059694.2016.57. PubMed PMID: 20608227.
- Nakahashi A, Miura N, Monde K, Tsukamoto S. Stereochemical studies of hexylitaconic acid, an inhibitor of p53-HDM2 interaction. Bioorganic & medicinal chemistry letters. 2009;19(11):3027-30. doi: 10.1016/j.bmcl.2009.04.057. PubMed PMID: 19414261.
- Bagal SK, Chapman ML, Marron BE, Prime R, Storer RI, Swain NA. Recent progress in sodium channel modulators for pain. Bioorganic & medicinal chemistry letters. 2014;24(16):3690-9. doi: 10.1016/j.bmcl.2014.06.038. PubMed PMID: 25060923.
- Katz E, Nisani S, Chamovitz DA. Indole-3-carbinol: a plant hormone combatting cancer. F1000Research. 2018;7. doi: 10.12688/f1000research.14127.1. PubMed PMID: 29904587; PubMed Central PMCID: PMC5989150.
- Brew CT, Aronchik I, Hsu JC, Sheen JH, Dickson RB, Bjeldanes LF, et al. Indole-3-carbinol activates the ATM signaling pathway independent of DNA damage to stabilize p53 and induce G1 arrest of human mammary epithelial cells. International journal of cancer Journal international du cancer. 2006;118(4):857-68. doi: 10.1002/ijc.21445. PubMed PMID: 16152627.
- Vogel SM, Bauer MR, Joerger AC, Wilcken R, Brandt T, Veprintsev DB, et al. Lithocholic acid is an endogenous inhibitor of MDM4 and MDM2. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(42):16906-10. doi: 10.1073/pnas.1215060109. PubMed PMID: 23035244; PubMed Central PMCID: PMC3479485.
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844-8. doi: 10.1126/science.1092472. PubMed PMID: 14704432.
- Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. Journal of medicinal chemistry. 2013;56(14):5979-83. doi: 10.1021/jm400487c. PubMed PMID: 23808545.
- Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer research. 2014;74(20):5855-65. doi: 10.1158/0008-5472.CAN-14-0799. PubMed PMID: 25145672; PubMed Central PMCID: PMC4247201.
- Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nature medicine. 2004;10(12):1321-8. doi: 10.1038/nm1146. PubMed PMID: 15558054.