Worldwide, the prevalence of surgery under general anesthesia has significantly increased, both because of modern anesthetic and pain-control techniques and because of better diagnosis and the increased complexity of surgical techniques. Apart from developing new concepts in the surgical field, researchers and clinicians are now working on minimizing the impact of surgical trauma and offering minimal invasive procedures due to the recent discoveries in the field of cellular and molecular mechanisms that have revealed a systemic inflammatory and pro-oxidative impact not only in the perioperative period but also in the long term, contributing to more difficult recovery, increased morbidity and mortality, and a negative financial impact. Detailed molecular and cellular analysis has shown an overproduction of inflammatory and pro-oxidative species, responsible for augmenting the systemic inflammatory status and making postoperative recovery more difficult. Moreover, there are a series of changes in certain epigenetic structures, the most important being the microRNAs.
- general anesthesia
- redox
- inflammation
- antioxidants
- hypermetabolism
- microRNAs
- oxidative stress
1. Introduction
2. Redox Disturbance and Inflammation during General Anesthesia and Surgery Procedures
This entry is adapted from the peer-reviewed paper 10.3390/cells11121880
References
- Kirov, K.; Motamed, C.; Ndoko, S.K.; Dhonneur, G. TOF Count at Corrugator Supercilii Reflects Abdominal Muscles Relaxation Better than at Adductor Pollicis. Br. J. Anaesth. 2007, 98, 611–614.
- Honing, M.; Martini, C.; van Velzen, M.; Niesters, M.; Dahan, A.; Boon, M. Cholinergic Chemotransmission and Anesthetic Drug Effects at the Carotid Bodies. Molecules 2020, 25, 5974.
- Duţu, M.; Ivaşcu, R.; Tudorache, O.; Morlova, D.; Stanca, A.; Negoiţă, S.; Corneci, D. Neuromuscular monitoring: An update. Rom. J. Anaesth. Intensive Care 2018, 25, 55–60.
- Funcke, S.; Saugel, B.; Koch, C.; Schulte, D.; Zajonz, T.; Sander, M.; Gratarola, A.; Ball, L.; Pelosi, P.; Spadaro, S.; et al. Individualized, Perioperative, Hemodynamic Goal-Directed Therapy in Major Abdominal Surgery (IPEGASUS Trial): Study Protocol for a Randomized Controlled Trial. Trials 2018, 19, 273.
- Bedreag, O.H.; Rogobete, A.F.; Sarandan, M.; Cradigati, A.C.; Papurica, M.; Dumbuleu, M.C.; Chira, A.M.; Rosu, O.M.; Sandesc, D. Oxidative Stress in Severe Pulmonary Trauma in Critical Ill Patients. Antioxidant Therapy in Patients with Multiple Trauma—A Review. Anestezjol. Intensywna Ter. 2015, 47, 351–359.
- Hsing, C.H.; Wang, J.J. Clinical implication of perioperative inflammatory cytokine alteration. Acta Anaesthesiol. Taiwan 2015, 53, 23–28.
- Boehm, O.; Baumgarten, G.; Hoeft, A. Epidemiology of the High-Risk Population: Perioperative Risk and Mortality after Surgery. Curr. Opin. Crit. Care 2015, 21, 322–327.
- Riedel, B.; Browne, K.; Silbert, B. Cerebral Protection: Inflammation, Endothelial Dysfunction, and Postoperative Cognitive Dysfunction. Curr. Opin. Anaesthesiol. 2014, 27, 89–97.
- Romagnoli, S.; Ricci, Z. Postoperative Acute Kidney Injury. Minerva Anestesiol. 2014, 81, 684–696.
- Snyder, G.L.; Greenberg, S. Effect of Anaesthetic Technique and Other Perioperative Factors on Cancer Recurrence. Br. J. Anaesth. 2010, 105, 106–115.
- Busti, A.J.; Hooper, J.S.; Amaya, C.J.; Kazi, S. Effects of Perioperative Antiinflammatory and Immunomodulating Therapy on Surgical Wound Healing. Pharmacotherapy 2005, 25, 1566–1591.
- Papurica, M.; Rogobete, A.F.; Sandesc, D.; Dumache, R.; Nartita, R.; Sarandan, M.; Cradigati, A.C.; Luca, L.; Vernic, C.; Bedreag, O.H. Redox Changes Induced by General Anesthesia in Critically Ill Patients with Multiple Traumas. Mol. Biol. Int. 2015, 2015, 238586.
- Denk, S.; Perl, M.; Huber-Lang, M. Damage- and Pathogen-Associated Molecular Patterns and Alarmins: Keys to Sepsis? Eur. Surg. Res. 2012, 48, 171–179.
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic Activators of Immune Responses. Curr. Opin. Immunol. 2005, 17, 359–365.
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a Clinical Response. J. Clin. Invest. 2012, 122, 2711–2719.
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of Chromatin Protein HMGB1 by Necrotic Cells Triggers Inflammation. Nature 2002, 418, 191–195.
- Tbahriti, H.F.; Meknassi, D.; Moussaoui, R.; Messaoudi, A.; Zemour, L.; Kaddous, A.; Bouchenak, M.; Mekki, K. Inflammatory Status in Chronic Renal Failure: The Role of Homocysteinemia and Pro-Inflammatory Cytokines. World J. Nephrol. 2013, 2, 31–37.
- Little, J.P.; Simtchouk, S.; Schindler, S.M.; Villanueva, E.B.; Gill, N.E.; Walker, D.G.; Wolthers, K.R.; Klegeris, A. Mitochondrial Transcription Factor A (Tfam) Is a pro-Inflammatory Extracellular Signaling Molecule Recognized by Brain Microglia. Mol. Cell. Neurosci. 2014, 60, 88–96.
- Kirchhoff, C.; Biberthaler, P.; Mutschler, W.E.; Faist, E.; Jochum, M.; Zedler, S. Early Down-Regulation of the pro-Inflammatory Potential of Monocytes Is Correlated to Organ Dysfunction in Patients after Severe Multiple Injury: A Cohort Study. Crit. Care 2009, 13, R88.
- Phillipson, M.; Kubes, P. The Neutrophil in Vascular Inflammation. Nat. Med. 2011, 17, 1381–1390.
- Reis, G.S.; Augusto, V.S.; Silveira, A.P.C.; Jordão, A.A.; Baddini-Martinez, J.; Poli Neto, O.; Rodrigues, A.J.; Evora, P.R.B. Oxidative-Stress Biomarkers in Patients with Pulmonary Hypertension. Pulmonary 2013, 3, 856–861.
- Hafner, S.; Radermacher, P.; Frick, M.; Dietl, P.; Calzia, E. Hyperglycemia, Oxidative Stress, and the Diaphragm: A Link between Chronic Co-Morbidity and Acute Stress? Crit. Care 2014, 18, 149.
- Cellular, O.S.; Overload, C. Sevoflurane Protects Ventricular Myocytes against Oxidative Stress-Induced Cellular Ca2+ Overload and Hypercontracture. Anesthesiology 2013, 119, 606–620.
- Breitenbach, M.; Rinnerthaler, M.; Weber, M.; Breitenbach-Koller, H.; Karl, T.; Cullen, P.; Basu, S.; Haskova, D.; Hasek, J. The Defense and Signaling Role of NADPH Oxidases in Eukaryotic Cells: Review. Wien. Med. Wochenschr. 2018, 168, 286–299.
- Ren, X.; Wang, M.; Wang, Y.; Huang, A. Superoxide anion generation response to wound in Arabidopsis hypocotyl cutting. Plant Signal Behav. 2021, 16, 1848086.
- Constantino, L.; Gonçalves, R.C.; Giombelli, V.R.; Tomasi, C.D.; Vuolo, F.; Kist, L.W.; Medeiros, G.; de Oliveira, T.; Augusto, M.; Pasquali, D.B.; et al. Regulation of Lung Oxidative Damage by Endogenous Superoxide Dismutase in Sepsis. Intensive Care Med. Exp. 2014, 2, 17.
- Milkovic, L.; Gasparovic, A.C.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793.
- Lv, P.; Xue, P.; Dong, J.; Peng, H.; Clewell, R.; Wang, A.; Wang, Y.; Peng, S.; Qu, W.; Zhang, Q.; et al. Keap1 Silencing Boosts Lipopolysaccharide-Induced Transcription of Interleukin 6 via Activation of Nuclear Factor ΚB in Macrophages. Toxicol. Appl. Pharmacol. 2013, 272, 697–702.
- Pagano, G.; Talamanca, A.A.; Castello, G.; Cordero, M.D.; Ischia, M.; Gadaleta, M.N.; Pallardó, F.V.; Petrović, S.; Tiano, L.; Zatterale, A. Oxidative Stress and Mitochondrial Dysfunction across Broad-Ranging Pathologies: Toward Mitochondria-Targeted Clinical Strategies. Oxidative Med. Cell. Longev. 2014, 2014, 541230.
- Waldbaum, S.; Patel, M. Mitochondria, Oxidative Stress, and Temporal Lobe Epilepsy. Epilepsy Res. 2010, 88, 23–45.
- Zang, Q.S.; Martinez, B.; Yao, X.; Maass, D.L.; Ma, L.; Wolf, S.E.; Minei, J.P. Sepsis-Induced Cardiac Mitochondrial Dysfunction Involves Altered Mitochondrial-Localization of Tyrosine Kinase Src and Tyrosine Phosphatase SHP2. PLoS ONE 2012, 7, e43424.
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial Dynamics and Mitochondrial Quality Control. Redox Biol. 2015, 4, 6–13.
- Kozlov, A.V.; Bahrami, S.; Calzia, E.; Dungel, P.; Gille, L.; Kuznetsov, A.V.; Troppmair, J. Mitochondrial Dysfunction and Biogenesis: Do ICU Patients Die from Mitochondrial Failure? Ann. Intensive Care 2011, 1, 41.
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial Reactive Oxygen Species: A Double Edged Sword in Ischemia/Reperfusion vs Preconditioning. Redox Biol. 2014, 2, 702–714.
- Sun, S.; Hu, F.; Wu, J.; Zhang, S. Cannabidiol Attenuates OGD/R-Induced Damage by Enhancing Mitochondrial Bioenergetics and Modulating Glucose Metabolism via Pentose-Phosphate Pathway in Hippocampal Neurons. Redox Biol. 2017, 11, 577–585.
- Gorelenkova Miller, O.; Behring, J.B.; Siedlak, S.L.; Jiang, S.; Matsui, R.; Bachschmid, M.M.; Zhu, X.; Mieyal, J.J. Upregulation of Glutaredoxin-1 Activates Microglia and Promotes Neurodegeneration: Implications for Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 967–982.
- Porfire, A.S.; Leucuţa, S.E.; Kiss, B.; Loghin, F.; Pârvu, A.E. Investigation into the Role of Cu/Zn-SOD Delivery System on Its Antioxidant and Antiinflammatory Activity in Rat Model of Peritonitis. Pharmacol. Rep. 2014, 66, 670–676.
- Edem, V.F.; Kosoko, A.; Akinyoola, S.B.; Owoeye, O.; Rahamon, S.K.; Arinola, O.G. Plasma Antioxidant Enzymes, Lipid Peroxidation and Hydrogen Peroxide in Wistar Rats Exposed to Dichlorvos Insecticide. Sch. Res. Libr. Arch. Appl. Sci. Res. 2012, 4, 1778–1781.
- Risnes, S.F.; Hartwig, A. Impact of Cadmium on Antioxidant Enzymes in HCT116 Cells and Protective Interaction by Selenium. Perspect. Sci. 2015, 3, 55.
- Samarghandian, S.; Afshari, R.; Farkhondeh, T. Effect of Long-Term Treatment of Morphine on Enzymes, Oxidative Stress Indices and Antioxidant Status in Male Rat Liver. Int. J. Clin. Exp. Med. 2014, 7, 1449–1453.
- Lee, B.-J.; Lin, J.-S.; Lin, Y.-C.; Lin, P.-T. Effects of L-Carnitine Supplementation on Oxidative Stress and Antioxidant Enzymes Activities in Patients with Coronary Artery Disease: A Randomized, Placebo-Controlled Trial. Nutr. J. 2014, 13, 79.
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Redox Biology Effects of N-Acetylcysteine, Oral Glutathione (GSH) and a Novel Sublingual Form of GSH on Oxidative Stress Markers: A Comparative Crossover Study. Redox Biol. 2015, 6, 198–205.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Babu, D.; Leclercq, G.; Goossens, V.; Vanden, T.; van Hamme, E.; Vandenabeele, P.; Lefebvre, R.A. Mitochondria and NADPH Oxidases Are the Major Sources of TNF-α/Cycloheximide-Induced Oxidative Stress in Murine Intestinal Epithelial MODE-K Cells. Cell. Signal. 2015, 27, 1141–1158.
- Jaganjac, M.; Milkovic, L.; Zarkovic, N.; Zarkovic, K. Oxidative Stress and Regeneration. Free Radic. Biol. Med. 2022, 181, 154–165.
- Li, P.; Stetler, R.A.; Leak, R.K.; Shi, Y.; Li, Y.; Yu, W.; Bennett, M.V.L.; Chen, J. Oxidative Stress and DNA Damage after Cerebral Ischemia: Potential Therapeutic Targets to Repair the Genome and Improve Stroke Recovery. Neuropharmacology 2018, 134, 208–217.
- Carcy, R.; Cougnon, M.; Poet, M.; Durandy, M.; Sicard, A.; Counillon, L.; Blondeau, N.; Hauet, T.; Tauc, M.; Pisani, D.F. Targeting Oxidative Stress, a Crucial Challenge in Renal Transplantation Outcome. Free Radic. Biol. Med. 2021, 169, 258–270.
- Yuan, Q.; Yuan, Y.; Zheng, Y.; Sheng, R.; Liu, L.; Xie, F.; Tan, J. Anti-Cerebral Ischemia Reperfusion Injury of Polysaccharides: A Review of the Mechanisms. Biomed. Pharmacother. 2021, 137, 111303.
- Padmavathi, G.; Ramkumar, K.M. MicroRNA Mediated Regulation of the Major Redox Homeostasis Switch, Nrf2, and Its Impact on Oxidative Stress-Induced Ischemic/Reperfusion Injury. Arch. Biochem. Biophys. 2021, 698, 108725.
- Hu, Y.; Deng, H.; Xu, S.; Zhang, J. MicroRNAs Regulate Mitochondrial Function in Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2015, 16, 24895–24917.
- Chi, O.Z.; Barsoum, S.; Rah, K.H.; Liu, X.; Weiss, H.R. Local O2 Balance in Cerebral Ischemia-Reperfusion Improved during Pentobarbital Compared with Isoflurane Anesthesia. J. Stroke Cerebrovasc. Dis. 2015, 24, 1196–1203.
- Zhai, F.; Zhang, X.; Guan, Y.; Yang, X.; Li, Y.; Song, G.; Guan, L. Expression Profiles of MicroRNAs after Focal Cerebral Ischemia/Reperfusion Injury in Rats. Neural Regen. Res. 2012, 7, 917–923.
- Freeman, L.R.; Keller, J.N. Oxidative Stress and Cerebral Endothelial Cells: Regulation of the Blood-Brain-Barrier and Antioxidant Based Interventions. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 822–829.
- Zhang, Z.; Yan, J.; Shi, H. Neurobiology of Disease Role of Hypoxia Inducible Factor 1 in Hyperglycemia-Exacerbated Blood-Brain Barrier Disruption in Ischemic Stroke. Neurobiol. Dis. 2016, 95, 82–92.
- Xia, H.; Cheng, Z.; Cheng, Y.; Xu, Y. Investigating the Passage of Tetramethylpyrazine-Loaded Liposomes across Blood-Brain Barrier Models in Vitro and Ex Vivo. Mater. Sci. Eng. C 2016, 69, 1010–1017.
- Robertson, C.S.; Narayan, R.K.; Gokaslan, Z.L.; Pahwa, R.; Grossman, R.G.; Caram, P.J.; Allen, E. Cerebral Arteriovenous Oxygen Difference as an Estimate of Cerebral Blood Flow in Comatose Patients. J. Neurosurg. 1989, 70, 222–230.
- Bouzat, P.; Sala, N.; Payen, J.-F.; Oddo, M. Beyond Intracranial Pressure: Optimization of Cerebral Blood Flow, Oxygen, and Substrate Delivery after Traumatic Brain Injury. Ann. Intensive Care 2013, 3, 23.
- Weiss, H.R.; Grayson, J.; Liu, X.; Barsoum, S.; Shah, H.; Chi, O.Z. Cerebral Ischemia and Reperfusion Increases the Heterogeneity of Local Oxygen Supply/Consumption Balance. Stroke 2013, 44, 2553–2558.
- Sakai, H.; Sheng, H.; Yates, R.B.; Ishida, K.; Pearlstein, R.D.; Warner, D.S. Isoflurane Provides Long-Term Protection against Focal Cerebral Ischemia in the Rat. Anesthesiology 2007, 106, 92–99.
- Gambim, M.; de Oliveira do Carmo, A.; Marti, L.; Veríssimo-Filho, S.; Lopes, L.; Janiszewski, M. Platelet-Derived Exosomes Induce Endothelial Cell Apoptosis through Peroxynitrite Generation: Experimental Evidence for a Novel Mechanism of Septic Vascular Dysfunction. Crit. Care 2007, 11, R107.
- Carnes, C.A.; Chung, M.K.; Nakayama, T.; Nakayama, H.; Baliga, R.S.; Piao, S.; Kanderian, A.; Pavia, S.; Hamlin, R.L.; McCarthy, P.M.; et al. Ascorbate Attenuates Atrial Pacing-Induced Peroxynitrite Formation and Electrical Remodeling and Decreases the Incidence of Postoperative Atrial Fibrillation. Circ. Res. 2001, 89, e32–e38.
- Erecinska, M.; Thoresen, M.; Silver, I.A. Effects of Hypothermia on Energy Metabolism in Mammalian Central Nervous System. J. Cereb. Blood Flow Metab. 2003, 23, 513–530.
- Scheufler, K.M.; Lehnert, A.; Rohrborn, H.J.; Nadstawek, J.; Thees, C. Individual Value of Brain Tissue Oxygen Pressure, Microvascular Oxygen Saturation, Cytochrome Redox Level, and Energy Metabolites in Detecting Critically Reduced Cerebral Energy State during Acute Changes in Global Cerebral Perfusion. J. Neurosurg. Anesthesiol. 2004, 16, 210–219.
- Antoniades, C.; Tousoulis, D.; Vasiliadou, C.; Pitsavos, C.; Toutouza, M.; Tentolouris, C.; Marinou, K.; Stefanadis, C. Genetic Polymorphisms G894T on the ENOS Gene Is Associated with Endothelial Function and VWF Levels in Premature Myocardial Infarction Survivors. Int. J. Cardiol. 2006, 107, 95–100.
- Abd-elbaset, M.; Arafa, E.A. Quercetin Modulates INOS, ENOS and NOSTRIN Expressions and Attenuates Oxidative Stress in Warm Hepatic Ischemia-Reperfusion Injury in Rats. Beni-Suef Univ. J. Basic Appl. Sci. 2015, 4, 246–255.
- Silver, J.H.; Jaffe, R.A.; López, J.R. Plasma Nitrite as an Indicator of Cerebral Ischemia during Extracranial/Intracranial Bypass Surgery in Moyamoya Patients. J. Stroke Cerebrovasc. Dis. 2020, 29, 104830.
- Suzuki, H.; Kanamaru, H.; Kawakita, F.; Asada, R.; Fujimoto, M.; Shiba, M. Cerebrovascular pathophysiology of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Histol. Histopathol. 2021, 36, 143–158.
- Washington, C.W.; Zipfel, G.J. Detection and Monitoring of Vasospasm and Delayed Cerebral Ischemia: A Review and Assessment of the Literature. Neurocrit. Care 2011, 15, 312–317.
- Liu, T.-J.; Zhang, J.-C.; Gao, X.-Z.; Tan, Z.-B.; Wang, J.-J.; Zhang, P.-P.; Cheng, A.-B.; Zhang, S.-B. Effect of Sevoflurane on the ATPase Activity of Hippocampal Neurons in a Rat Model of Cerebral Ischemia-Reperfusion Injury via the CAMP-PKA Signaling Pathway. Kaohsiung J. Med. Sci. 2018, 34, 22–33.
- Coles, J.P.; Fryer, T.D.; Smielewski, P.; Chatfield, D.A.; Steiner, L.A.; Johnston, A.J.; Downey, S.P.; Williams, G.B.; Aigbirhio, F.; Hutchinson, P.J.; et al. Incidence and Mechanisms of Cerebral Ischemia in Early Clinical Head Injury. J. Cereb. Blood Flow Metab. 2004, 24, 202–211.
- Drake, C.; Boutin, H.; Jones, M.S.; Denes, A.; Mccoll, B.W.; Selvarajah, J.R.; Hulme, S.; Georgiou, R.F.; Hinz, R.; Gerhard, A.; et al. Brain, Behavior, and Immunity Brain Inflammation Is Induced by Co-Morbidities and Risk Factors for Stroke. Brain Behav. Immun. 2011, 25, 1113–1122.
- Hartley, O.; Offord, R.E. Engineering Chemokines to Develop Optimized HIV Inhibitors. Curr. Protein Pept. Sci. 2005, 6, 207–219.
- Stamatovic, S.M.; Phillips, C.M.; Martinez-Revollar, G.; Keep, R.F.; Andjelkovic, A.V. Involvement of Epigenetic Mechanisms and Non-Coding RNAs in Blood-Brain Barrier and Neurovascular Unit Injury and Recovery after Stroke. Front. Neurosci. 2019, 13, 864.
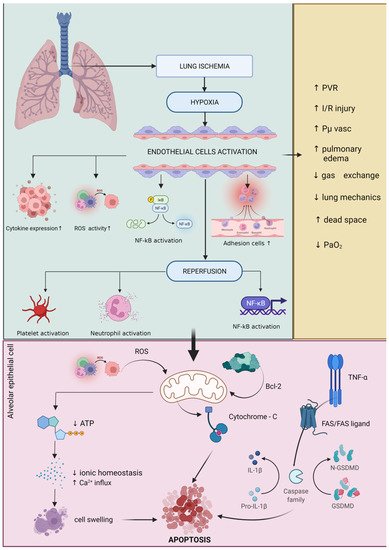
- De Perrot, M.; Liu, M.; Waddell, T.K.; Keshavjee, S. Ischemia-Reperfusion-Induced Lung Injury. Am. J. Respir. Crit. Care Med. 2003, 167, 490–511.
- Liu, X.; Feng, Z.; Du, L.; Huang, Y.; Ge, J.; Deng, Y.; Mei, Z. The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury. Int. J. Mol. Sci. 2020, 21, 120.
- Stoica, L.; Dobrescu, A.; Isaic, A.; Verdeæ, G.; Taråa, C.; Lazãr, F. Metabolic and Hormonal Changes after Sleeve Gastrectomy and Mini Gastric Bypass in a Rat Model of Induced Type 2 Diabetes Mellitus and Obesity. Chirurgia 2019, 114, 732–738.
- Rogobete, A.F.; Grintescu, I.M.; Bratu, T.; Bedreag, O.H.; Papurica, M.; Crainiceanu, Z.P.; Popovici, S.E.; Sandesc, D. Assessment of Metabolic and Nutritional Imbalance in Mechanically Ventilated Multiple Trauma Patients: From Molecular to Clinical Outcomes. Diagnostics 2019, 9, 171.
- Eppinger, M.J.; Deeb, G.M.; Bolling, S.F.; Ward, P.A. Mediators of ischemia-reperfusion injury of rat lung. Am. J. Pathol. 1997, 150, 1773–1784.
- Qayumi, A.K.; Jamieson, W.R.E.; Godin, D.V.; Lam, S.; Ko, K.M.; Germann, E.; van den Broek, J. Response to Allopurinol Pretreatment in a Swine Model of Heart-Lung Transplantation. J. Investig. Surg. 1990, 3, 331–340.
- Chen, Q.; Xu, J.; Li, L.; Li, H.; Mao, S.; Zhang, F.; Zen, K.; Zhang, C.; Zhang, Q. MicroRNA-23a/b and MicroRNA-27a/b Suppress Apaf-1 Protein and Alleviate Hypoxia-Induced Neuronal Apoptosis. Cell Death Dis. 2014, 5, e1132.
- Danial, N.N.; Korsmeyer, S.J. Cell Death: Critical Control Points. Cell 2004, 116, 205–219.
- Borutaite, V.; Jekabsone, A.; Morkuniene, R.; Brown, G.C. Inhibition of Mitochondrial Permeability Transition Prevents Mitochondrial Dysfunction, Cytochrome c Release and Apoptosis Induced by Heart Ischemia. J. Mol. Cell. Cardiol. 2003, 35, 357–366.
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344.
- Cobelens, P.M.; van Putte, B.P.; Kavelaars, A.; Heijnen, C.J.; Kesecioglu, J. Inflammatory Consequences of Lung Ischemia-Reperfusion Injury and Low-Pressure Ventilation. J. Surg. Res. 2008, 153, 295–301.
- Hickey, M.J.; Sharkey, K.A.; Sihota, E.G.; Reinhardt, P.H.; Macmicking, J.D.; Nathan, C.; Kubes, P. Inducible Nitric Oxide Synthase-Deficient Mice Have Enhanced Leukocyte-Endothelium Interactions in Endotoxemia. FASEB J. 1997, 11, 955–964.
- Ovechkin, A.V.; Lominadze, D.; Sedoris, K.C.; Robinson, T.W.; Tyagi, S.C.; Roberts, A.M. Lung Ischemia-Reperfusion Injury: Implications of Oxidative Stress and Platelet-Arteriolar Wall Interactions. Arch. Physiol. Biochem. 2007, 113, 1–12.
- Saelens, X.; Festjens, N.; van de Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic Proteins Released from Mitochondria in Cell Death. Oncogene 2004, 23, 2861–2874.
- Scarabelli, T.M.; Stephanou, A.; Pasini, E.; Comini, L.; Raddino, R.; Knight, R.A.; Latchman, D.S. Different Signaling Pathways Induce Apoptosis in Endothelial Cells and Cardiac Myocytes during Ischemia/Reperfusion Injury. Circ. Res. 2002, 90, 745–748.
- Zhang, X.; Lemasters, J.J. Free Radical Biology and Medicine Translocation of Iron from Lysosomes to Mitochondria during Ischemia Predisposes to Injury after Reperfusion in Rat Hepatocytes. Free Radic. Biol. Med. 2013, 63, 243–253.
- Mikawa, K.; Akamatsu, H.; Nishina, K.; Shiga, M.; Maekawa, N.; Obara, H.; Niwa, Y. Propofol Inhibits Human Neutrophil Functions. Anesth. Analg. 1998, 87, 695–700.
- Ohmizo, H.; Iwama, H.; Sugita, T. Complement Activation by Propofol and Its Effect during Propofol Anaesthesia. Anaesth. Intensive Care 1999, 27, 623–627.
- Hoff, G.; Bauer, I.; Larsen, B.; Bauer, M. Modulation of Endotoxin-Stimulated TNF-Alpha Gene Expression by Ketamine and Propofol in Cultured Human Whole Blood. Anaesthesist 2001, 50, 494–499.
- Mitsuhata, H.; Shimizu, R.; Yokoyama, M.M. Suppressive Effects of Volatile Anesthetics on Cytokine Release in Human Peripheral Blood Mononuclear Cells. Int. J. Immunopharmacol. 1995, 17, 529–534.
- Giraud, O.; Molliex, S.; Rolland, C.; Leçon-Malas, V.; Desmonts, J.-M.; Aubier, M.; Dehoux, M. Halogenated Anesthetics Reduce Interleukin-1beta-Induced Cytokine Secretion by Rat Alveolar Type II Cells in Primary Culture. Anesthesiology 2003, 98, 74–81.
- Kotani, N.; Takahashi, S.; Sessler, D.I.; Hashiba, E.; Kubota, T.; Hashimoto, H.; Matsuki, A. Volatile Anesthetics Augment Expression of Proinflammatory Cytokines in Rat Alveolar Macrophages during Mechanical Ventilation. Anesthesiology 1999, 91, 187–197.
- Zhang, L.; Zhang, J.; Yang, L.; Dong, Y.; Zhang, Y.; Xie, Z. Isoflurane and Sevoflurane Increase Interleukin-6 Levels through the Nuclear Factor-Kappa B Pathway in Neuroglioma Cells. Br. J. Anaesth. 2013, 110 (Suppl. S1), i82–i91.
- Hudetz, J.A.; Gandhi, S.D.; Iqbal, Z.; Patterson, K.M.; Pagel, P.S. Elevated Postoperative Inflammatory Biomarkers Are Associated with Short- and Medium-Term Cognitive Dysfunction after Coronary Artery Surgery. J. Anesth. 2011, 25, 1–9.
- Manfreda, S.E.; Dunzendorfer, S.; Schratzberger, P.; Buratti, T.; Reinisch, N.; Kähler, C.M.; List, W.F.; Wiedermann, C.J. The Chemotaxis of Human Peripheral Blood B Lymphocytes by Beta-Endorphin Is Reversible by Naloxone. Anesth. Analg. 1998, 86, 670–672.
- Yeager, M.P.; Yu, C.T.; Campbell, A.S.; Moschella, M.; Guyre, P.M. Effect of Morphine and Beta-Endorphin on Human Fc Receptor-Dependent and Natural Killer Cell Functions. Clin. Immunol. Immunopathol. 1992, 62, 336–343.
- Baek, S.B.; Shin, M.S.; Han, J.H.; Moon, S.W.; Chang, B.; Jeon, J.W.; Yi, J.W.; Chung, J.Y. Rocuronium Bromide Inhibits Inflammation and Pain by Suppressing Nitric Oxide Production and Enhancing Prostaglandin E(2) Synthesis in Endothelial Cells. Int. Neurourol. J. 2016, 20, 296–303.
- Bargellini, A.; Rovesti, S.; Barbieri, A.; Vivoli, R.; Roncaglia, R.; Righi, E.; Borella, P. Effects of Chronic Exposure to Anaesthetic Gases on Some Immune Parameters. Sci. Total Environ. 2001, 270, 149–156.
- Yamamoto, S.; Niida, S.; Azuma, E.; Yanagibashi, T.; Muramatsu, M.; Huang, T.T.; Sagara, H.; Higaki, S.; Ikutani, M.; Nagai, Y.; et al. Inflammation-Induced Endothelial Cell-Derived Extracellular Vesicles Modulate the Cellular Status of Pericytes. Sci. Rep. 2015, 5, 8505.
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-ΚB, Inflammation, and Metabolic Disease. Cell Metab. 2011, 13, 11–22.
- Liu, J.; Xie, B.; Chen, S.; Jiang, F.; Meng, W. Association Study of Two Inflammation-Related Polymorphisms with Susceptibility to Hepatocellular Carcinoma: A Meta-Analysis. BMC Med. Genet. 2014, 15, 92.
- Papurica, M.; Sandesc, D.; Rogobete, A.F.; Nartita, R.; Vernic, C.; Popovici, S.E.; Bedreag, O.H. Cardioprotective Effects Induced by Preconditioning with Halogenated Anesthetics. J. Interdiscip. Med. 2016, 1, 23–31.
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding Cellular Redox Homeostasis: A Challenge for Precision Medicine. Int. J. Mol. Sci. 2022, 23, 106.
- Bedreag, O.H.; Rogobete, A.F.; Cradigati, C.A.; Sarandan, M.; Nartita, R.; Horhat, F.G.; Popovici, S.E.; Sandesc, D.; Papurica, M. A Novel Evaluation of Microvascular Damage in Critically Ill Polytrauma Patients by Using Circulating MicroRNAs. Rev. Română Med. Lab. 2016, 24, 21–30.
- Corcoran, T.B.; Engel, A.; Sakamoto, H.; O’Shea, A.; O’Callaghan-Enright, S.; Shorten, G.D. The Effects of Propofol on Neutrophil Function, Lipid Peroxidation and Inflammatory Response during Elective Coronary Artery Bypass Grafting in Patients with Impaired Ventricular Function. Br. J. Anaesth. 2006, 97, 825–831.
- Potočnik, I.; Janković, V.N.; Šostarič, M.; Jerin, A.; Štupnik, T.; Skitek, M.; Markovič-Božič, J.; Klokočovnik, T. Antiinflammatory Effect of Sevoflurane in Open Lung Surgery with One-Lung Ventilation. Croat. Med. J. 2014, 55, 628–637.
- Markovic-Bozic, J.; Karpe, B.; Potocnik, I.; Jerin, A.; Vranic, A.; Novak-Jankovic, V. Effect of Propofol and Sevoflurane on the Inflammatory Response of Patients Undergoing Craniotomy. BMC Anesthesiol. 2016, 16, 18.
- Roh, G.U.; Song, Y.; Park, J.; Ki, Y.M.; Han, D.W. Effects of Propofol on the Inflammatory Response during Robot-Assisted Laparoscopic Radical Prostatectomy: A Prospective Randomized Controlled Study. Sci. Rep. 2019, 9, 5242.
- Wijeysundera, D.N.; Committee, E.R.; Duncan, D.; Nkonde-price, C.; Virani, S.S.; Washam, J.B.; Fleischmann, K.E.; Vice, P.G.; Fleisher, L.A.; Guideline, P.; et al. Perioperative Beta Blockade in Noncardiac Surgery: A Systematic Review for the 2014 ACC/AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery. Circulation 2014, 64, 2246–2264.
- Julier, K.; da Silva, R.; Garcia, C.; Bestmann, L.; Frascarolo, P.; Zollinger, A.; Chassot, P.-G.; Schmid, E.R.; Turina, M.I.; von Segesser, L.K.; et al. Preconditioning by Sevoflurane Decreases Biochemical Markers for Myocardial and Renal Dysfunction in Coronary Artery Bypass Graft Surgery: A Double-Blinded, Placebo-Controlled, Multicenter Study. Anesthesiology 2003, 98, 1315–1327.
- Fukazawa, K.; Lee, H.T. Volatile Anesthetics and AKI: Risks, Mechanisms, and a Potential Therapeutic Window. J. Am. Soc. Nephrol. 2014, 25, 884–892.
- Cai, J.; Xu, R.; Yu, X.; Fang, Y.; Ding, X. Volatile Anesthetics in Preventing Acute Kidney Injury after Cardiac Surgery: A Systematic Review and Meta-Analysis. J. Thorac. Cardiovasc. Surg. 2014, 148, 3127–3136.
- Kempson, S.A.; Zhou, Y.; Danbolt, N.C. The Betaine/GABA Transporter and Betaine: Roles in Brain, Kidney, and Liver. Front. Physiol. 2014, 5, 159.
- Bagley, E.E. Opioid and GABAB Receptors Differentially Couple to an Adenylyl Cyclase/Protein Kinase a Downstream Effector after Chronic Morphine Treatment. Front. Pharmacol. 2014, 5, 148.
- Adermark, L.; Söderpalm, B.; Burkhardt, J.M. Brain Region Specific Modulation of Ethanol-Induced Depression of GABAergic Neurons in the Brain Reward System by the Nicotine Receptor Antagonist Mecamylamine. Alcohol 2014, 48, 455–461.
- Wagner, J.; Strosing, K.M.; Spassov, S.G.; Lin, Z.; Engelstaedter, H.; Tacke, S.; Hoetzel, A.; Faller, S. Sevoflurane Posttreatment Prevents Oxidative and Inflammatory Injury in Ventilator-Induced Lung Injury. PLoS ONE 2018, 13, e0192896.