Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Self-healing polymers are categorized as smart materials that are capable of surface protection and prevention of structural failure. Polyurethane/polyurea, as one of the representative coatings, has also attracted attention for industrial applications.
- polyurea coating
- self-healing
- self-healing mechanism
- formulation optimization
1. Introduction
Many times, structures that require protection are in poorly accessible areas, which makes maintenance work difficult. As an industrial class of smart materials, self-healing polymers are distinguished by the fact that they can repair themselves without detection or manual intervention [1]. The concept of self-healing in polymers was introduced in the 1980s [2]. Interest in the self-healing of polymers grew when significant research progress was made by Sottos [3] in 1993 and White et al. [4] in 2001. Eventually, the European Space Agency [5] and the US Air Force [6] saw the advantages and made investments for further advances in self-healing polymers. In 2007, the first international conference on self-healing materials was held [7][8]. In recent years, studies have been conducted to equip self-healing polyurethane and polyurea with adequate mechanical properties by changing the bonds present in the polymers. The goal is to achieve an optimal balance between self-healing efficiency and mechanical properties. Lee [9] added azomethine groups to polyurethane and obtained a self-healing efficiency of 86% and a tensile strength of 50 MPa. Qian [10] introduced alkyl diselenide to polyurethane and managed to recover 100% of the polymer’s initial mechanical properties [11]. Without the need for detection or manual intervention, this can ensure that protective coatings are capable of providing continuous protection against corrosion for a prolonged time, even in the absence of short-windowed routine checks. This can also reduce the high cost of corrosion and maintenance work in poorly accessible areas. With such benefits provided by self-healing polymers, it is anticipated that the self-healing market will continue to expand significantly until at least 2025 [12].
Both polyurea and polyurethane are commonly found in coatings that protect against moisture, corrosion, abrasion, and chemicals. However, there are some differences between polyurea and polyurethane that make one a better choice over the other in different applications. Polyurea is the product of the reaction between polyamine and isocyanate (R-NCO + R’-NH2 → R-NH-CO–NH–R’), while polyurethane is the product of the reaction between polyol, isocyanate, and a catalyst (R-NCO + H2O → R-NH2 + CO2 & R-NCO + R’-NH2 → R-NH-CO–NH–R) [3][12]. Comparing polyurethane and polyurea, polyurea has higher tensile strength and a shorter curing time. Despite these superior properties, research in recent years has mainly focused only on the self-healing of polyurethane. Lee [9] and Hu et al. [13] used azomethine diols to achieve self-healing polyurethane elastomers with tensile strengths of at least 40 MPa. Li et al. [14] and Wang et al. [15] incorporated the Diels–Alder structure to achieve self-healing polyurethane with up to 95% healing efficiency [16]. Only limited work conducted is related to the self-healing of polyurea, and the frequency of conducting maintenance work can be greatly reduced, which makes it more capable of protecting existing structures against weathering effects [17][18] and corrosion [19]. Polyurea coatings are also known to have good corrosion resistance against seawater [20] and are capable of providing both underwater and on-land blast resistance [21] to underlying structures.
There are also varieties of formulation possibilities, incorporated with various additives, to achieve the desired performance. Much like that for polyurethane chemistry, these formulation possibilities are made possible by the selection of different types of raw materials. Thus, the selection of appropriate raw materials for the polyurea coating system can be a very complex procedure. The formulation consists of two main components (Part A: isocyanate component, reactive diluent; Part B: polyether amines, chain extenders, and additives and pigments). The most commonly used Part A components are toluene-2,4-diisocyanate (TDI) and diphenylmethane diisocyanate (MDI). TDI prepolymers with NCO content of 45 wt.% to 55 wt.% or MDI prepolymers with NCO content of 14 wt.% to 17 wt.% are preferred for standard preparations of polyurea spray coatings. In contrast, Part B, which is also named Part R, the resin blend component or polyether part, is normally a mixture of amine-terminated ethylene oxide and/or propylene oxide polyether with molecular weights varying from 200 to 5000 g/mole. Polyether amines are mainly used to increase flexibility, toughness, hydrophilicity, or hydrophobicity, and they also offer various reactivities, good thermal stabilities, colorlessness, and low viscosity.
2. Self-Healing Mechanisms of Polyurea
Figure 1 shows the performance of original material and self-healing material over time. Performance is defined as the original property of the material, be it tensile strength, hardness, or corrosion resistance. Service lifetime is defined as the period of time in which the material is above its limit of reliability and in working condition before it needs to be replaced. Curve (a) represents original material with a decrease in performance over time and a limited-service lifetime. Curve (b) represents traditionally improved material with a slight extension of the service lifetime. Curve (c) represents self-healing material. When there is damage inflicted on the material, its performance drops. After healing, the performance increases again and decreases due to wear over time. Once there is more damage, the cycle repeats. Due to the ability to enhance performance after healing, self-healing material is able to extend the service lifetime to a greater extent.
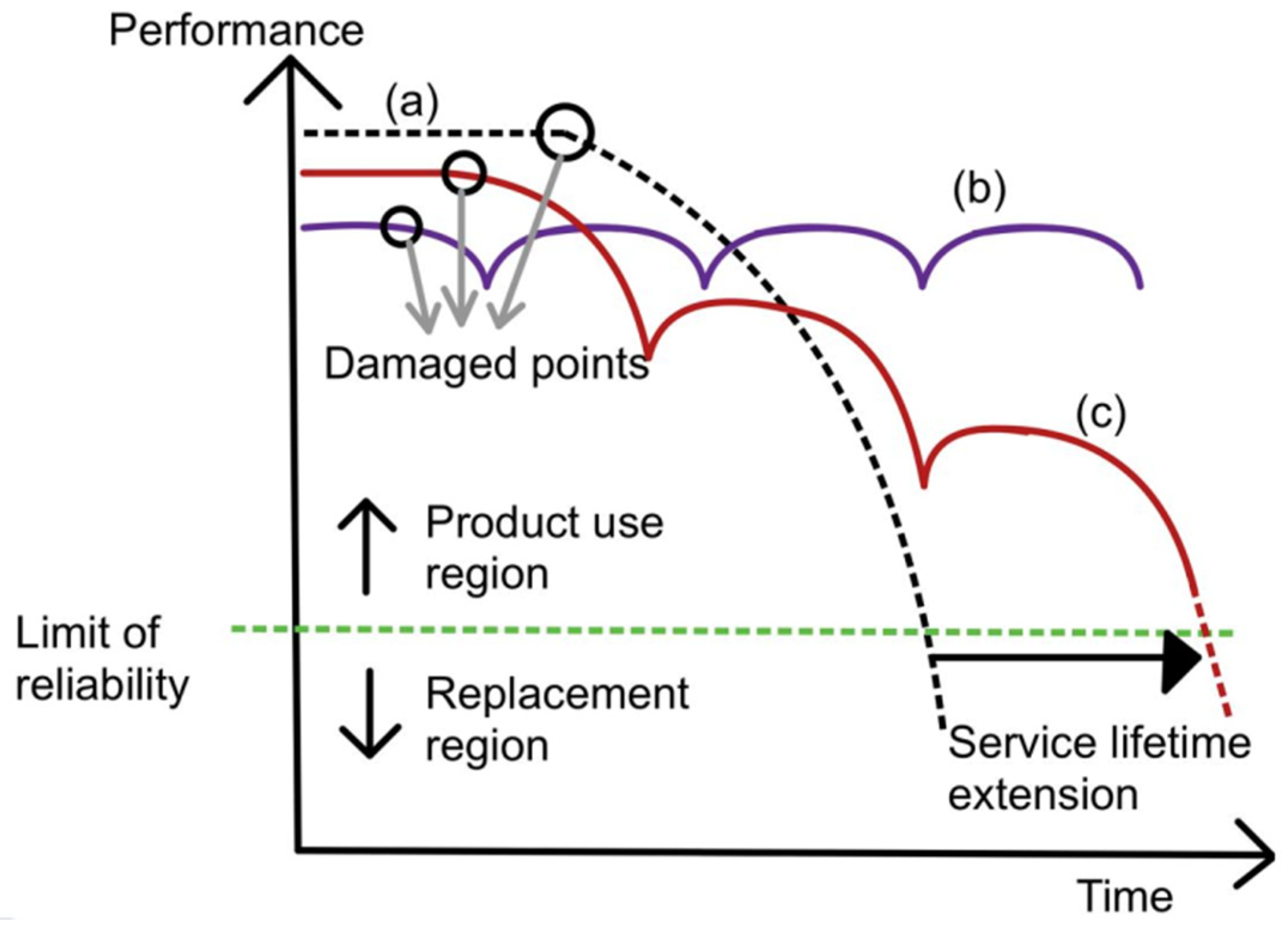 Figure 1. Graph of performance plotted against time for curve (a) (normal material), curve (b) (ideal self-healing material), and curve (c) (self-healing material) [22].
Figure 1. Graph of performance plotted against time for curve (a) (normal material), curve (b) (ideal self-healing material), and curve (c) (self-healing material) [22].The working principle of polyurea self-healing mechanisms is to fill cracks by introducing more healing components, which can polymerize and seal damage in the material. Alternatively, it can also be addressed by encouraging continuous chemical reactions, which can form bonds to close gaps between the separated faces of material due to the damage.
There are multiple mechanisms by which self-healing can occur in polyurea. Self-healing can occur by including healing agents, which are encapsulated and embedded in the polymeric matrix [4][23]. The healing agents will then be released when the encapsulation is broken. Besides that, self-healing can also occur via molecular interdiffusion [24][25], supramolecular non-covalent interaction [26], or dynamic covalent bonding [15][27]. The mechanisms of self-healing polyurea can then be differentiated into extrinsic mechanisms and intrinsic mechanisms, or autonomous mechanisms and non-autonomous mechanisms. Extrinsic and intrinsic refer to the use of additional healing agents to achieve self-healing [28]. Extrinsic mechanisms use additional healing agents, while intrinsic mechanisms do not. Autonomous and non-autonomous refer to the use of external stimuli that are not generated by damage to the material [29][30]. Autonomous mechanisms do not require the use of external stimuli for self-healing, while non-autonomous mechanisms require stimuli such as heat and light. In this research, researchers differentiate the mechanisms into extrinsic and intrinsic. The categorization of mechanisms is illustrated in Figure 2.
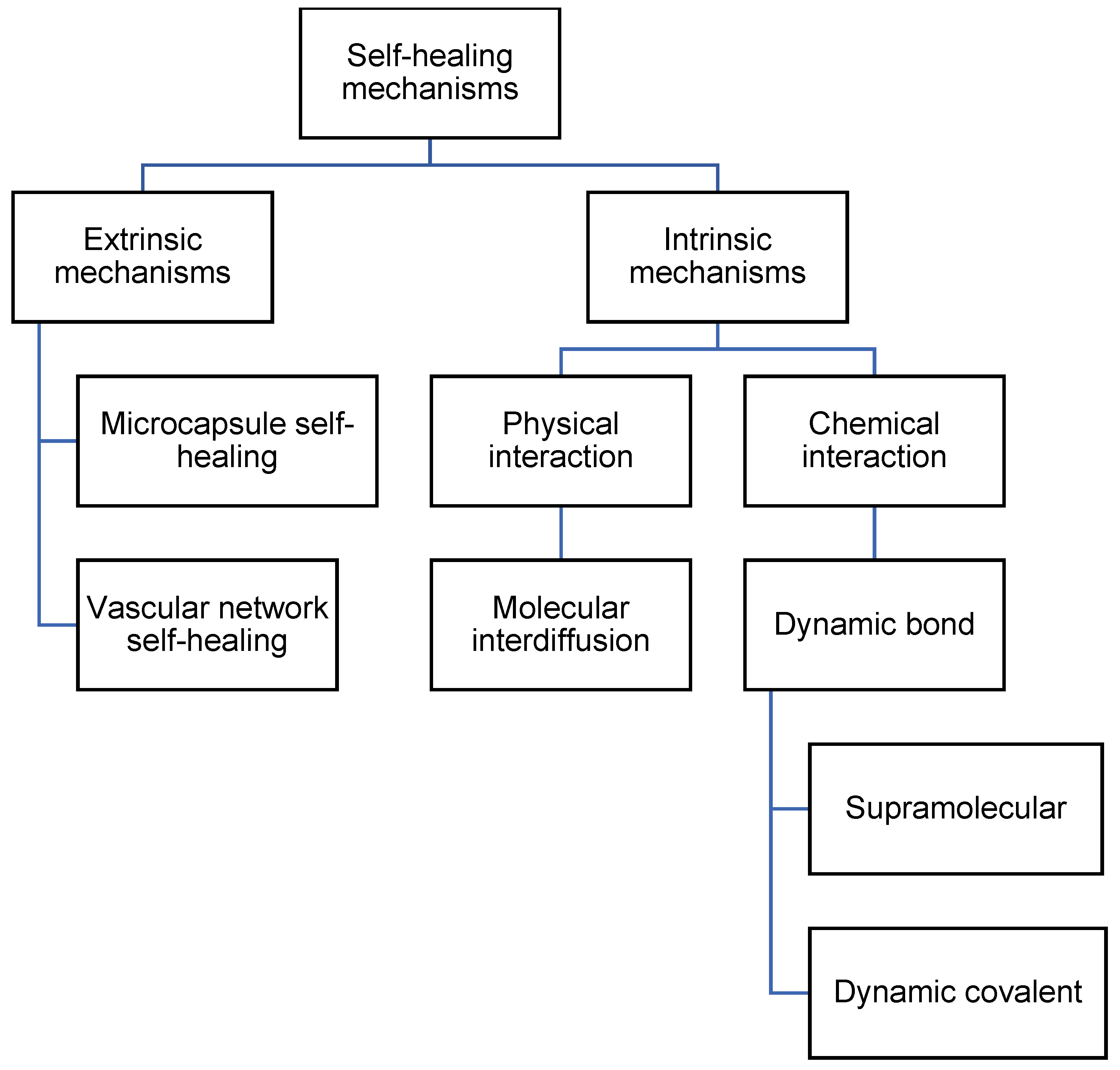 Figure 2. Flowchart listing the types of self-healing mechanisms in polyurea.
Figure 2. Flowchart listing the types of self-healing mechanisms in polyurea.2.1. Extrinsic Mechanisms
Extrinsic mechanisms are defined as self-healing processes that make use of different types of containers holding prefilled healing agents that are then embedded in the matrix. Extrinsic mechanisms include microcapsule self-healing and vascular network self-healing.
2.1.1. Microcapsule Self-Healing
The microcapsule self-healing approach involves the use of healing agents that are encapsulated in capsules to heal cracks in the material. White et al. [4] prepared urea–formaldehyde microcapsules with sizes of 50–200 micrometers that are filled with dicyclopentadiene (DCPD). These DCPD-filled microcapsules are then embedded in the matrix together with Grubb’s catalyst. During service time, cracks form in the matrix. As the cracks propagate, the microcapsules rupture and release their contents. The DCPC flows out of the capsules onto the matrix. Eventually, when DCPD interacts with Grubb’s catalyst, polymerization takes place and fills the crack. Hence, self-healing occurs. This process is illustrated in Figure 3.
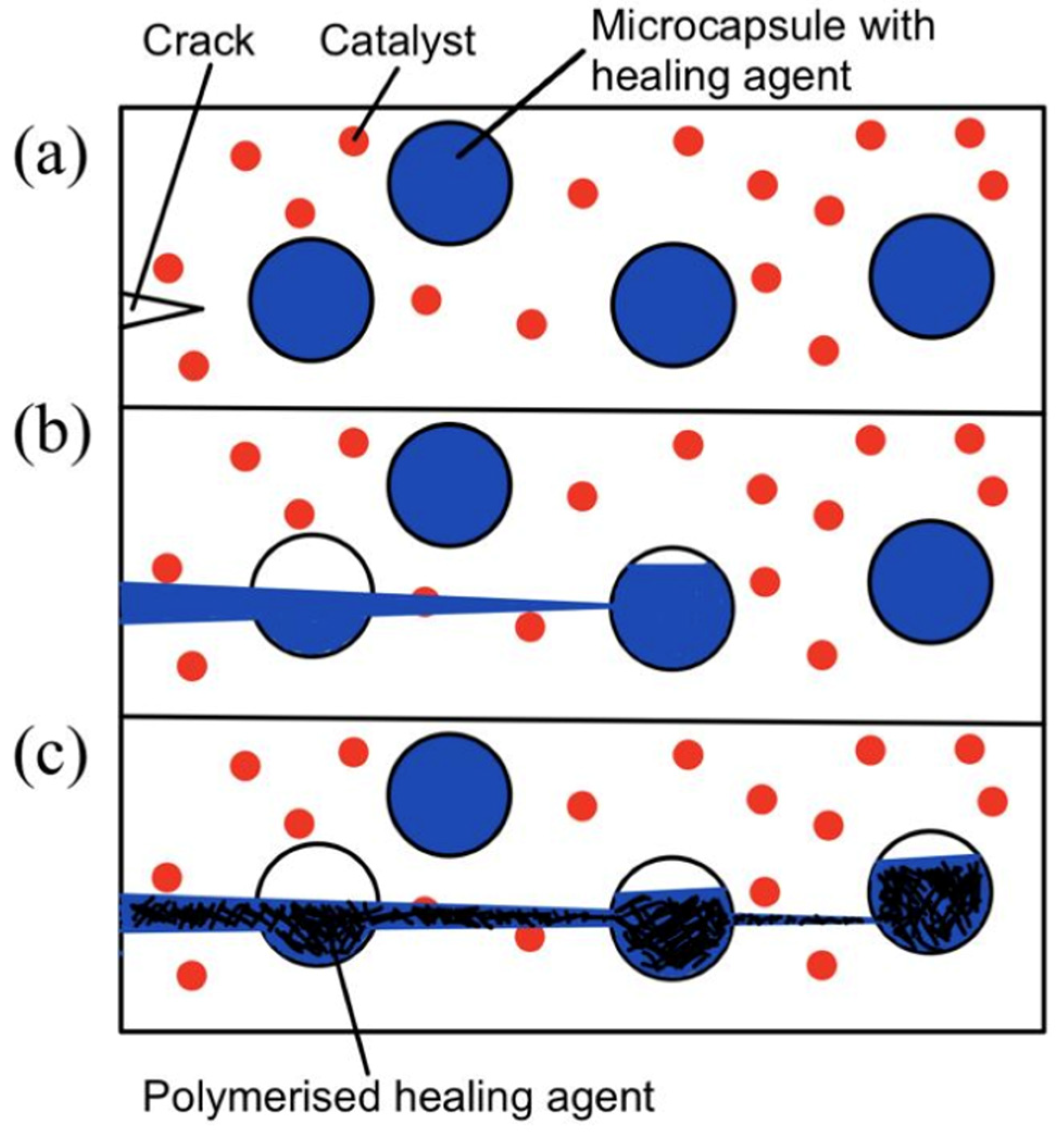 Figure 3. (a) Crack nucleation occurs in the matrix, (b) crack propagation takes place and ruptures microcapsules, and (c) microcapsules’ contents flow out and polymerize upon contact with the catalyst [4].
Figure 3. (a) Crack nucleation occurs in the matrix, (b) crack propagation takes place and ruptures microcapsules, and (c) microcapsules’ contents flow out and polymerize upon contact with the catalyst [4].In recent years, multiple studies have been conducted based on this working principle [31]. However, moisture inevitably penetrates the intact coating and reacts with the healing agent. To solve the problem of the loss of healing agents due to moisture penetration, Sun et al. [32] used a double-wall microcapsule. The inner wall formed by the reaction between Tetraethylenepentamine (TEPA) and isocyanate and the outer wall formed by a physical unclonable function (PUF) constituted double-walled microcapsules. This double-wall microcapsule is proven to exhibit better water resistance and enhance long-term anticorrosion performance.
With advances in research, applications that use polyurea microcapsules have become increasingly diverse [33]. Polyurea microcapsules can be used in various areas, from self-healing anticorrosion coatings [34][35] to energy storage [36][37].
2.1.2. Vascular Network Self-Healing
Unlike the microcapsule self-healing approach, the healing agents in a vascular network self-healing system are not stored in capsules. The healing agents in a biomimetic vascular network [38] are stored in microchannels that resemble blood vessels in the human body. This vascular network self-healing approach was first demonstrated by C. Dry [23][24]. The basic working principle behind vascular network self-healing is illustrated in Figure 4.
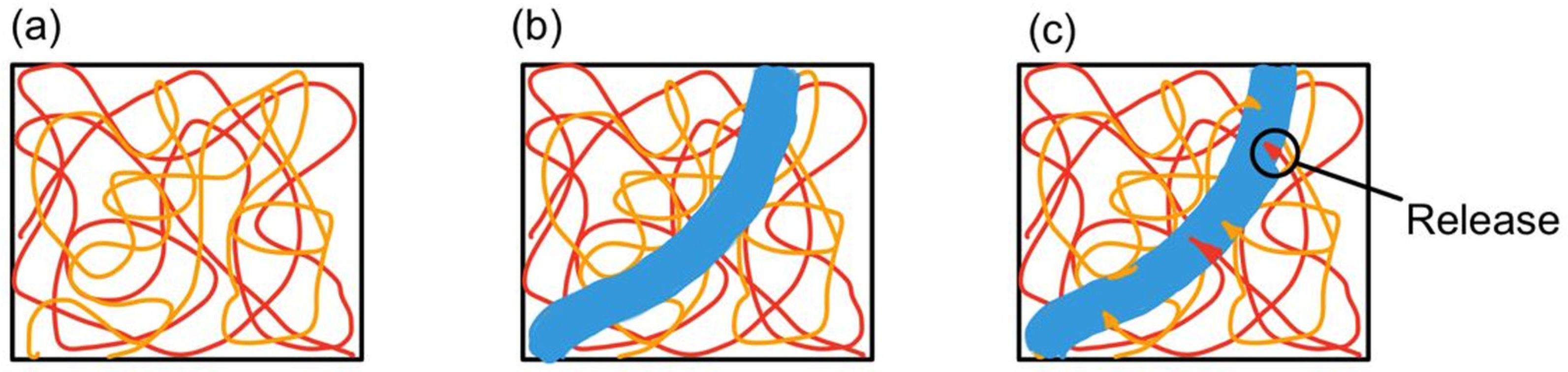 Figure 4. (a) Vascular network embedded in polymer composite matrix, (b) a cut is made (blue region), and (c) monomers from microchannel leak into the matrix [39].
Figure 4. (a) Vascular network embedded in polymer composite matrix, (b) a cut is made (blue region), and (c) monomers from microchannel leak into the matrix [39].Polymeric microchannels and a catalyst are embedded in the matrix. The monomer is then forced to infiltrate into the microchannel. Once the microchannel is damaged by crack propagation, the monomer leaks into the matrix and comes into contact with the surrounding catalyst [40]. As a result, polymerization occurs and fills the crack. Hence, self-healing takes place.
Based on the way in which microchannels are embedded into the matrix, the vascular network self-healing system can be categorized into one-dimensional, two-dimensional, and three-dimensional. A one-dimensional vascular network system means that the polymer composite matrix consists of one-dimensional pipelines (Figure 5a). Both the resin and hardener are encapsulated in two different one-dimensional pipelines. Two-dimensional and three-dimensional vascular network systems have interconnected microvascular structures with both resin and hardener pipelines flowing in two dimensions (Figure 5b) and three dimensions (Figure 5c). With two- and three-dimensional microvascular structures, multiple healing cycles are made possible, as there is a constant flow of healing agents.
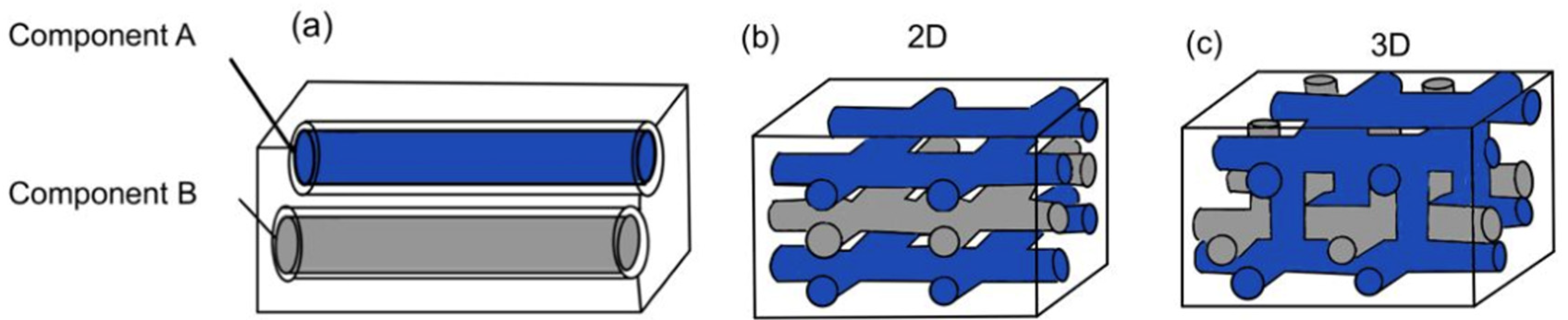 Figure 5. (a) One-dimensional vascular network system, (b) two-dimensional vascular network system, and (c) three-dimensional vascular network system [28].
Figure 5. (a) One-dimensional vascular network system, (b) two-dimensional vascular network system, and (c) three-dimensional vascular network system [28].However, the ability of this vascular network to achieve multiple healing cycles is largely dependent on the architecture of the network [41]. As healing agents leak out from the pipeline, thrombosis of the network tends to occur. This blocks any further flow of healing agent from the pipeline to the damage site. Hence, the self-healing capability is hindered. To design a better network architecture for the enhancement of self-healing, recent studies have been tapping into additive manufacturing (AM) techniques [42][43]. AM methods such as 3D printing have been employed to produce sacrificial scaffolds to template the healing system.
2.2. Intrinsic Mechanisms
Intrinsic mechanisms are defined as self-healing processes that take place without the need for a healing agent or catalyst. These mechanisms are based on the inherent reversibility of the chemical bonds present in the polymeric matrix, which can be rearranged [28][39]. Intrinsic mechanisms are further divided into physical interaction and chemical interaction. With physical interaction, there is molecular interdiffusion. With chemical interaction, there is dynamic bonding.
2.2.1. Physical Interaction
The phenomenon of molecular interdiffusion was first discovered when two pieces of the same polymer were brought together at temperatures above their glass transition temperatures [44]. The interfaces between the two pieces disappear, and the mechanical strength at the interface increases [44][45]. As a result, extensive research was conducted around the 1980s [25][46][47]. In particular, the Wool and O’Connor model [25] to explain the process of crack healing was more widespread. In the 1990s, research in this area slowed down [44]. The crack-healing process consists of five stages, as summarized in Table 1.
| Stages | Definition |
|---|---|
| 1. Surface rearrangement | The roughness or topography of the surface changes with external factors (pressure, time, and temperature). |
| 2. Surface approach | Healing can only occur when surfaces are brought together, or a gap is filled with healing fluid. |
| 3. Wetting | Surfaces have to wet each other and form an interface before healing can occur. |
| 4. Diffusion | The most critical stage of strength development. |
| 5. Randomization | Refers to the equilibration of the non-equilibrium conformations of chains near the surfaces. |
Surface rearrangement is the first step for self-healing to take place via physical interaction. Self-healing needs to take place for crack healing to occur. When the fractured surface is in contact with the healing agent, its surface topography will change with time, temperature, and pressure. This occurs due to the diffusion of chain-end distributions. The chain end can be designed accordingly to increase the efficiency of crack healing [49]. For instance, lower-surface-tension parts are applied with chain ends, and low-molecular-weight species can allow faster diffusion of chain ends from the bulk to the surface for reaction with the healing agent to heal the crack. However, oxidation and cross-linking can chemically react, disrupting diffusion kinetics. This could prevent self-healing and eventually prevent crack healing [16].
The second step is surface approach. Surface approach is the most essential step in self-healing. Surface approach must occur for self-healing [42]. The fractured surfaces have to be either brought together or surrounded by the healing agent. After the fractured surfaces approach each other, they have to wet each other to form an interface between them. The concept of wetting and the spreading of fluid was explored by Brochard [50]. As some fluids are better at wetting than others, the wettability of surfaces can determine the efficiency of self-healing [49]. The wetting stage ensures that the material has high enough chain mobility to promote diffusion in Stage 4 [42]. In a case in which the fractured surfaces have undergone chemical reactions such as oxidation, the fractured surfaces will no longer be wettable by fluid and hence will disrupt the process of self-healing.
Diffusion is the fourth stage of crack healing mechanisms. Diffusion of the chains in the polymeric matrix leads to the entanglement of polymer chains. This stage promotes the recovery of the mechanical properties of the healed material [42]. The entanglement of mobile polymer chains near the surface is also a random motion that occurs during diffusion. Hence, it is important that surface rearrangement has taken place so that chain ends are near the surface. Then, surface approach and wetting allow for diffusion across the fractured surfaces and interpenetration into the unruptured matrix material.
Randomization is the last stage of the crack healing mechanisms explained by Wool and O’Connor. This randomization stage refers to the equilibration of the non-equilibrium conformations of chains near the fractured surfaces [51]. It is also the stage where the weight distribution and orientation of chain segments near the fractured surfaces are restored.
2.2.2. Chemical Interaction
Dynamic bonds refer to any type of bonds capable of undergoing repetitive breaking and reformation at an equilibrium rate [52]. Dynamic bonds can be further split into supramolecular and dynamic covalent self-healing. Supramolecular self-healing can take place at equilibrium, while dynamic covalent self-healing requires an additional intervention, such as heat or UV [28].
- (a)
-
Supramolecular
In supramolecular self-healing, non-covalent bonds and transient bonds, such as hydrogen bonds, pi–pi stacking, and metal–ligand coordination bonds, are used to generate the network. This network can then undergo repetitive breaking and reformation, allowing multiple healing events. Hydrogen bonds are capable of associating and dissociating spontaneously under ambient conditions [53]. This makes hydrogen bonds suitable for use in intrinsic self-healing materials. A hydrogen-bond-based self-healing polymer was first developed by Leibler [54], who used fatty acids and urea to design and synthesize molecules that would cross-link via hydrogen bonds. Leibler discovered that the broken samples were able to heal at a room temperature of 20 degrees Celsius until scars were invisible [55]. Upon repeated breaking and healing, the repaired sample will still break along the scar location unless a longer healing duration is permitted. As such, the sample can be broken and healed via repeated hydrogen bonding. Comparing monodentate and bidentate urea hydrogen bonds, bidentate hydrogen bonds are capable of forming stronger hydrogen linkages [56] and hence dissociate close to the polymer decomposition temperature [57].
- (b)
-
Dynamic Covalent
In dynamic covalent self-healing, covalent bonds such as disulfide bonds, Diels–Alder reactions, and imine bonds are used. Figure 6 shows the reversibility of disulfide bonds [58] and their three-step exchange mechanism [59]. Firstly, the thiol is ionized to form a thiolate anion via initialization progress. Secondly, the sulfur atom of the disulfide undergoes nucleophilic attack by the thiolate anion through propagation. This causes the cleavage of the original S-S bond and the formation of another. Thirdly, protonation may form thiol via termination. Hence, another disulfide bond is formed again.
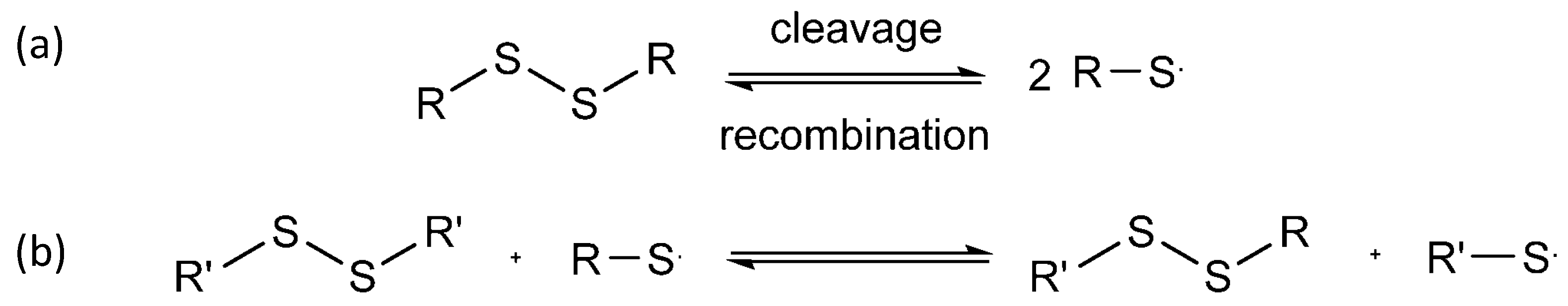 Figure 6. (a) Cleavage and recombination of disulfide bond; (b) 3 steps in disulfide exchange mechanism.
Figure 6. (a) Cleavage and recombination of disulfide bond; (b) 3 steps in disulfide exchange mechanism.2.3. Comparison between Extrinsic and Intrinsic Mechanisms
A brief summary and challenges faced when using extrinsic and intrinsic mechanisms are listed in Table 2.
| Advantages | Disadvantages | |
|---|---|---|
| Microcapsules (Extrinsic) |
|
|
| Vascular network (Extrinsic) |
|
|
| Dynamic bonds (Intrinsic) |
|
|
Generally, microcapsules (extrinsic) face the limitation of being single-use. The vascular network (extrinsic) faces the potential blockage of core fibers due to its network, which compromises its healing properties. On the other hand, dynamic bonds (intrinsic) are capable of infinite healing cycles. This research aims to find the optimum formulation of self-healing polyurea that is capable of healing even after multiple cuts. This is to meet the practical needs of industrial uses. Hence, this research focuses on intrinsic self-healing mechanisms.
This entry is adapted from the peer-reviewed paper 10.3390/polym14142808
References
- Li, Y.; Liu, Y.; Yao, B.; Narasimalu, S.; Dong, Z. Rapid preparation and antimicrobial activity of polyurea coatings with RE-Doped nano-ZnO. Microb. Biotechnol. 2022, 15, 548–560.
- Li, Y.; Fang, C.; Zhuang, W.-Q.; Wang, H.; Wang, X. Antimicrobial enhancement via Cerium (II)/Lanthanum (III)-doped TiO2 for emergency leak sealing polyurea coating system. npj Mater. Degrad. 2022, 6, 41.
- Rong, Z.; Li, Y.; Lim, R.Z.; Wang, H.; Dong, Z.; Li, K.; Wang, X. Fire-retardant effect of titania-polyurea coating and additional enhancement via aromatic diamine and modified melamine polyphosphate. NPJ Mater. Degrad. 2022, 6, 38.
- White, S.R.; Sottos, N.R.; Geubelle, P.H.; Moore, J.S.; Kessler, M.R.; Sriram, S.R.; Brown, E.N.; Viswanathan, S. Autonomic healing of polymer composites. Nature 2001, 409, 794–797.
- ESA, Enabling Self-Healing Capabilities—A Small Step to Bio-Mimetic Materials, 4476, 2006, Issue 1. Available online: http://esamultimedia.esa.int/docs/gsp/materials_report_4476.pdf (accessed on 1 March 2007).
- Carlson, H.C.; Goretta, K. Basic materials research programs at the U.S. Air Force Office of Scientific Research. Mater. Sci. Eng. B 2006, 132, 2–7.
- Schmets, A.J.M.; Zwaag, S.v.d. International Conference on Self Healing. In Proceedings of the First International Conference on Self Healing Materials, Noordwijk aan Zee, The Netherlands, 18–20 April 2007.
- Asnaashari, M.; Grafton, R.J.; Johnnie, M. Precast Concrete Design-Construction of San Mateo-Hayward Bridge Widening Project. PCI J. 2005, 50, 26–43.
- Polyurea Market Size, Share & Trends Analysis Report By Raw Material, by Product (Coating, Lining, Adhesives & Sealants), by Application (Construction, Industrial, Transportation), and Segment Forecasts, 2019–2025. Grand View Research. Available online: https://www.grandviewresearch.com/industry-analysis/polyurea-market (accessed on 5 September 2019).
- Zechel, S.; Geitner, R.; Abend, M.; Siegmann, M.; Enke, M.; Kuhl, N.; Klein, M.; Vitz, J.; Gräfe, S.; Dietzek, B.; et al. Intrinsic self-healing polymers with a high E-modulus based on dynamic reversible urea bonds. NPG Asia Mater. 2017, 9, e420.
- Lee, D.-W.; Kim, H.-N.; Lee, D.-S. Design of Azomethine Diols for Efficient Self-Healing of Strong Polyurethane Elas-tomers. Molecules 2018, 23, 2928.
- Qian, Y.; An, X.; Huang, X.; Pan, X.; Zhu, J.; Zhu, X. Recyclable Self-Healing Polyurethane Cross-Linked by Alkyl Diselenide with Enhanced Mechanical Properties. Polymers 2019, 11, 773.
- Hu, J.; Mo, R.; Sheng, X.; Zhang, X. A self-healing polyurethane elastomer with excellent mechanical properties based on phase-locked dynamic imine bonds. Polym. Chem. 2020, 11, 2585–2594.
- Li, Y.; Yang, Z.; Zhang, J.; Ding, L.; Pan, L.; Huang, C.; Zheng, X.; Zeng, C.; Lin, C. Novel polyurethane with high self-healing efficiency for functional energetic composites. Polym. Test. 2019, 76, 82–89.
- Wang, Z.; Yang, H.; Fairbanks, B.D.; Liang, H.; Ke, J.; Zhu, C. Fast self-healing engineered by UV-curable polyurethane contained Diels-Alder structure. Prog. Org. Coatings 2019, 131, 131–136.
- Jiang, L.; Liu, Z.; Lei, Y.; Yuan, Y.; Wu, B.; Lei, J. Sustainable Thermosetting Polyurea Vitrimers Based on a Catalyst-Free Process with Reprocessability, Permanent Shape Reconfiguration and Self-Healing Performance. ACS Appl. Polym. Mater. 2019, 1, 3261–3268.
- Xu, J.; Chen, P.; Wu, J.; Hu, P.; Fu, Y.; Jiang, W.; Fu, J. Notch-Insensitive, Ultrastretchable, Efficient Self-Healing Supramolecular Polymers Constructed from Multiphase Active Hydrogen Bonds for Electronic Applications. Chem. Mater. 2019, 31, 7951–7961.
- Guan, S.W. 100% solids polyurethane and polyurea coatings technology. Coat. World 2003, 04, 49–58.
- Iqbal, N.; Sharma, P.; Kumar, D.; Roy, P. Protective polyurea coatings for enhanced blast survivability of concrete. Constr. Build. Mater. 2018, 175, 682–690.
- Nagaraj, S.; Babu, S.K. Protective polyurea coating for enhanced corrosion resistance of sole bars in railway coaches. Mater. Today Proc. 2019, 27, 2407–2411.
- Ping, L.; Jing, L.; Mingliang, M.; Yilong, S. Research on Seawater Corrosion Resistance of Spray Polyurea Protective Coating. Mater. Sci. Eng. 2018, 436, 012017.
- Dai, L.-H.; Wu, C.; An, F.-J.; Liao, S.-S. Experimental Investigation of Polyurea-Coated Steel Plates at Underwater Explosive Loading. Adv. Mater. Sci. Eng. 2018, 2018, 1264276.
- Garcia, S.J. Effect of polymer architecture on the intrinsic self-healing character of polymers. Eur. Polym. J. 2014, 53, 118–125.
- Dry, C. PASSIVE TUNEABLE FIBERS AND MATRICES. Int. J. Mod. Phys. B 1992, 6, 2763–2771.
- de Gennes, P.G. Reptation of a Polymer Chain in the Presence of Fixed Obstacles. J. Chem. Phys. 1971, 55, 572–579.
- Wool, R.P.; O’Connor, K.M. A theory crack healing in polymers. J. Appl. Phys. 1981, 52, 5953–5963.
- Li, T.; Zhang, C.; Xie, Z.; Xu, J.; Guo, B.-H. A multi-scale investigation on effects of hydrogen bonding on micro-structure and macro-properties in a polyurea. Polymer 2018, 145, 261–271.
- Chen, T.; Fang, L.; Li, X.; Gao, D.; Lu, C.; Xu, Z. Self-healing polymer coatings of polyurea-urethane/epoxy blends with reversible and dynamic bonds. Prog. Org. Coatings 2020, 147, 105876.
- Hia, I.L.; Vahedi, V.; Pasbakhsh, P. Self-Healing Polymer Composites: Prospects, Challenges, and Applications. Polym. Rev. 2016, 56, 225–261.
- Willocq, B.; Odent, J.; Dubois, P.; Raquez, J.-M. Advances in intrinsic self-healing polyurethanes and related composites. RSC Adv. 2020, 10, 13766–13782.
- Zhang, F.; Ju, P.; Pan, M.; Zhang, D.; Huang, Y.; Li, G.; Li, X. Self-healing mechanisms in smart protective coatings: A review. Corros. Sci. 2018, 144, 74–88.
- Ullah, H.; Azizli, K.A.M.; Man, Z.B.; Ismail, M.B.C.; Khan, M.I. The Potential of Microencapsulated Self-healing Materials for Microcracks Recovery in Self-healing Composite Systems: A Review. Polym. Rev. 2016, 56, 429–485.
- Sun, D.; Zhang, H.; Tang, X.-Z.; Yang, J. Water resistant reactive microcapsules for self-healing coatings in harsh environments. Polymer 2016, 91, 33–40.
- Thorne, M.F.; Simkovic, F.; Slater, A.G. Production of monodisperse polyurea microcapsules using microfluidics. Sci. Rep. 2019, 9, 17983.
- Gite, V.V.; Tatiya, P.D.; Marathe, R.J.; Mahulikar, P.P.; Hundiwale, D.G. Microencapsulation of quinoline as a corrosion inhibitor in polyurea microcapsules for application in anticorrosive PU coatings. Prog. Org. Coat. 2015, 83, 11–18.
- Njoku, C.N.; Bai, W.; Arukalam, I.O.; Yang, L.; Hou, B.; Njoku, D.I.; Li, Y. Epoxy-based smart coating with self-repairing polyurea-formaldehyde microcapsules for anticorrosion protection of aluminum alloy AA2024. J. Coatings Technol. Res. 2020, 17, 797–813.
- Zhou, J.; Xu, W.; Wang, Y.-N.; Shi, B. Preparation of polyurea microcapsules containing phase change materials in a rotating packed bed. RSC Adv. 2017, 7, 21196–21204.
- Guang-Long, Z.; Xiao-Zheng, L.; Zhi-Cheng, T.; Li-Xian, S.; Tao, Z. Microencapsulation of n-Hexadecane as a Phase Change Material in Polyurea. Acta Phys. -Chim. Sin. 2004, 20, 90–93.
- Williams, H.; Trask, R.; Knights, A.; Bond, I. Biomimetic reliability strategies for self-healing vascular networks in engineering materials. J. R. Soc. Interface 2008, 5, 735–747.
- An, S.; Lee, M.W.; Yarin, A.L.; Yoon, S.S. A review on corrosion-protective extrinsic self-healing: Comparison of microcapsule-based systems and those based on core-shell vascular networks. Chem. Eng. J. 2018, 344, 206–220.
- Toohey, K.S.; Sottos, N.R.; Lewis, J.A.; Moore, J.; White, S. Self-healing materials with microvascular networks. Nat. Mater. 2007, 6, 581–585.
- Qamar, I.P.S.; Sottos, N.R.; Trask, R.S. Grand challenges in the design and manufacture of vascular self-healing. Multifunct. Mater. 2020, 3, 013001.
- Almutairi, M.D.; Aria, A.I.; Thakur, V.K.; Khan, M.A. Self-Healing Mechanisms for 3D-Printed Polymeric Structures: From Lab to Reality. Polymers 2020, 12, 1534.
- Sanders, P.; Young, A.; Qin, Y.; Fancey, K.S.; Reithofer, M.R.; Guillet-Nicolas, R.; Kleitz, F.; Pamme, N.; Chin, J.M. Stereolithographic 3D printing of extrinsically self-healing composites. Sci. Rep. 2019, 9, 388.
- Wu, D.Y.; Meure, S.; Solomon, D. Self-healing polymeric materials: A review of recent developments. Prog. Polym. Sci. 2008, 33, 479–522.
- Billiet, S.; Hillewaere, X.K.D.; Teixeira, R.F.A.; Du Prez, F.E. Chemistry of Crosslinking Processes for Self-Healing Polymers. Macromol. Rapid Commun. 2013, 34, 290–309.
- Doi, M.; Edwards, S.F. Dynamics of concentrated polymer systems. Part 1. —Brownian motion in the equilibrium state. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1789–1801.
- Prager, S.; Tirrell, M. The healing process at polymer–Polymer interfaces. J. Chem. Phys. 1981, 75, 5194–5198.
- Kim, S.-M.; Jeon, H.; Shin, S.-H.; Park, S.-A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior Toughness and Fast Self-Healing at Room Temperature Engineered by Transparent Elastomers. Adv. Mater. 2018, 30, 1705145.
- Wool, R.P. Self-healing materials: A review. Soft Matter 2008, 4, 400–418.
- Brochard, F. Spreading of liquid drops on thin cylinders: The ‘‘manchon/droplet’’ transition. J. Chem. Phys. 1986, 84, 4664–4672.
- Wool, R.P. Chapter 8—Diffusion and autohesion. In Adhesion Science and Engineering; Dillard, D.A., Pocius, A.V., Chaudhury, M., Eds.; Elsevier Science B.V: Amsterdam, The Netherland, 2002; pp. 351–401.
- Wojtecki, R.J.; Meador, M.A.; Rowan, S. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 2011, 10, 14–27.
- He, M.; Chen, X.; Liu, D.; Wei, D. Two-dimensional self-healing hydrogen-bond-based supramolecular polymer film. Chin. Chem. Lett. 2019, 30, 961–965.
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980.
- Ionita, D.; Gaina, C.; Cristea, M.; Banabic, D. Tailoring the hard domain cohesiveness in polyurethanes by interplay between the functionality and the content of chain extender. RSC Adv. 2015, 5, 76852–76861.
- Tahir, M.; Heinrich, G.; Mahmood, N.; Boldt, R.; Wießner, S.; Stöckelhuber, K.W. Blending In Situ Polyurethane-Urea with Different Kinds of Rubber: Performance and Compatibility Aspects. Materials 2018, 11, 2175.
- Nevejans, S.; Ballard, N.; Miranda, J.I.; Reck, B.; Asua, J.M. The underlying mechanisms for self-healing of poly(disulfide)s. Phys. Chem. Chem. Phys. 2016, 18, 27577–27583.
- Black, S.P.; Sanders, J.K.M.; Stefankiewicz, A.R. Disulfide exchange: Exposing supramolecular reactivity through dynamic covalent chemistry. Chem. Soc. Rev. 2014, 43, 1861–1872.
This entry is offline, you can click here to edit this entry!