Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Diabetes mellitus, commonly referred to only as diabetes, is a group of metabolic disorders characterized by hyperglycemia over long periods of time. Neutrophils are the most abundant leukocytes in human blood, the primary effector cells of acute inflammation and the first responders to infections. Neutrophils are involved in obesity-related diabetes complications.
- neutrophils
- diabetes
- inflammation
1. Neutrophils in Type 1 Diabetes (T1D)
Diabetes mellitus, commonly referred to only as diabetes, is a group of metabolic disorders characterized by hyperglycemia over long periods of time. Diabetes is due to either lack of insulin secretion from the β cells in the pancreas or insulin resistance, a condition in which cells of the body do not respond properly to insulin [1]. There are two main types of diabetes: type 1 (insulin deficiency) and type 2 (insulin resistance) [2]. Both diabetes types are associated with serious clinical complications such as cardiovascular disorders, heart failure, atherosclerosis, diabetic neuropathy, diabetic retinopathy, and diabetic kidney disease [3][4][5].
Type 1 diabetes mellitus (T1D) is considered a T cell-mediated autoimmune disease, in which autoreactive T lymphocytes destroy the insulin-producing β cells in the pancreatic islets [6]. Much progress has been made in understanding T1D thanks to the nonobese diabetic (NOD) mouse animal model [7]. Similar to human T1D, NOD mice exhibit an autoimmune response towards β cells, resulting in the dysfunction and destruction of these cells. However, a limitation of this model is that in NOD mice, the initial antigen is insulin [8], while in humans, anti-islet autoantibodies are the most frequently detected autoantibodies [9]. In a study of Japanese T1D patients, it was reported that the main antigens recognized by autoantibodies were glutamic acid decarboxylase (GAD), insulinoma-associated antigen-2 (IA-2), zinc transporter 8 (ZnT8), and insulin [10].
Pioneering work with NOD mice demonstrated that physiological β cell death induced the recruitment and activation of B-1a cells and plasmacytoid dendritic cells to the pancreas [11]. These events represent the initial immunological steps for developing T1D. Importantly, in this first study, a significant early infiltration of neutrophils to the pancreas was also reported [11]. At the same time, it was found that circulating neutrophil numbers were reduced in T1D patients and that neutrophils were present in the pancreas of patients with T1D but not in patients with Neutrophils in Type 2 Diabetes (T2D) or in nondiabetic controls [12]. A reduction in circulating neutrophils has been confirmed in other studies and it is considered a hallmark of T1D [13][14][15][16][17]. Moreover, this reduction of neutrophils correlates with lower serum levels of neutrophil elastase (NE) and proteinase 3 (PR3) [18] and with faster disease progression [15][19]. The reduction of circulating neutrophils is due mainly to neutrophil infiltration into the pancreatic tissue [12][15]. In neonatal NOD mice, neutrophil infiltration [20][21] and NE concentrations in the pancreas [20][22] are already higher than in control mice as early as at two weeks of age. Macrophages and β cells produce chemokines CXCL1 and CXCL2, which in turn recruit CXCR2-expressing neutrophils to the pancreas [21] (Figure 1). Thus, neutrophils emerge as important cells participating in the early stages of T1D development. This is not too surprising, since recently neutrophils have been recognized as key components of both the innate and adaptive immune systems [23] and as important participants in the immunization and the effector phases of autoimmune diseases [24].
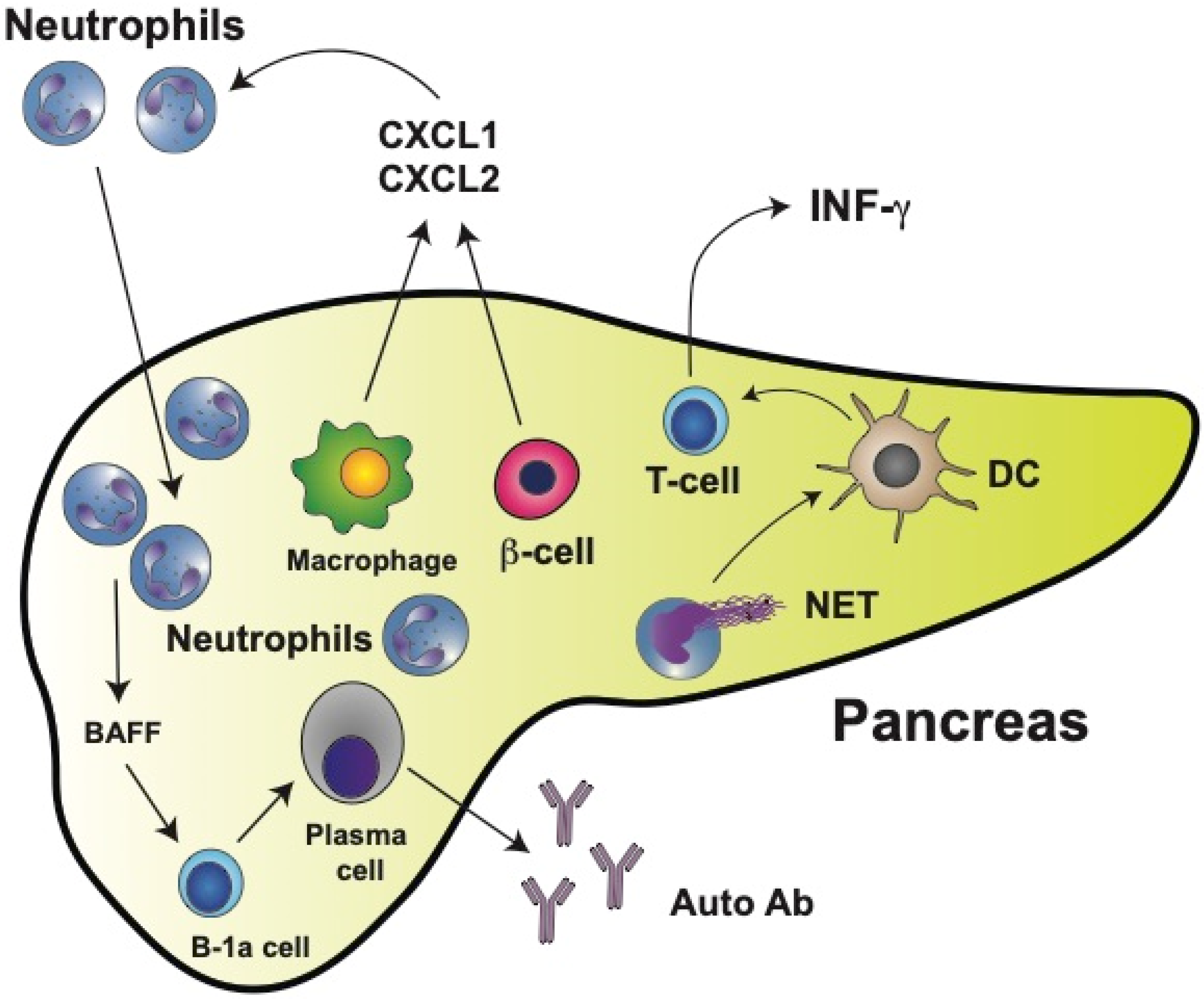
Figure 1. Type 1 diabetes mellitus (T1D) is characterized by neutrophil infiltration in the pancreas. In the pancreas, macrophages and insulin-producing β cells release chemokines CXCL1 and CXCL2, which in turn recruit neutrophils to the pancreas. Neutrophils can then directly activate B-1a cells via secreted cytokines such as BAFF (B cell-activating factor of the TNF family). BAFF is one of the main prosurvival factors for B cells as well as for antibody-producing plasma cells. Most plasma cells activated this way in the pancreas produce autoreactive antibodies (Auto Ab). The main antigens recognized by autoantibodies are glutamic acid decarboxylase (GAD), insulinoma-associated antigen-2 (IA-2), zinc transporter 8 (ZnT8), and insulin. Many neutrophils also release neutrophil extracellular traps (NET), which can activate dendritic cells (DCs). Activated DCs then stimulate T cells, leading to the production of interferon gamma (IFN-γ).
As seen in several autoimmune diseases, NETosis might contribute to promoting both inflammation and tissue damage. In the case of T1D, neutrophil extracellular traps (NET) components have been detected in circulation. However, there are contradictory reports. Increased NET components (NE and PR3 proteins) were reported in the serum of patients with T1D [15][25]. Yet, in a previous report, reduced circulating levels of NET components were found to correlate with the reduced number of circulating neutrophils [18]. However, NETosis within the pancreas clearly contributes to disease progression [26]. Neutrophil infiltration into pancreatic islets of NOD mice correlates with higher levels of citrullination [27]. Thus, by inhibiting NE with sivelestat or elafin [20] or PAD4 with BB-Cl-amidine [27], the development of diabetes was prevented in NOD mice. Similarly, by degrading NET with staphylococcal nuclease (SNase) (delivered to NOD mice by oral administration of modified Lactococcus lactis), pancreatic inflammation was reduced, β cell numbers increased, and glucose tolerance was improved [28]. Moreover, neutrophils isolated from T1D patients had an increased expression of PAD4 and showed enhanced NETosis after stimulation [29]. In vitro, NET isolated from T1D pediatric patients induced monocyte-derived dendritic cell activation, leading to the production of interferon gamma (IFN-γ) by T cells [30] (Figure 1). Although NET do not seem to directly induce the production of autoantibodies, they favor β cell damage resulting in exposure of the antigens recognized by anti-islet autoantibodies.
Neutrophils can also directly activate B cells via secreted cytokines such as BAFF (B cell-activating factor of the TNF family). BAFF, acting through its receptor [31], is one of the main prosurvival factors for B cells as well as for antibody-producing plasma cells [32] (Figure 1). Hence, neutrophils and NET components have an evident contribution to the development of T1D. These findings may open new opportunities for innovative therapeutic approaches in the future.
In addition, several neutrophil functions, including phagocytosis, degranulation, and production of reactive oxygen species (ROS), have been reported to be reduced in patients with T1D [33][34][35]. All these defects are thought to be caused by hyperglycemia [36][37]. However, it was recently found that in vitro neutrophil migration was impaired in neutrophils from T1D but not from T2D patients [16]. This functional defect was associated with the expression of L-selectin (CD62L) but not with high glucose concentrations [16]. Thus, it may be possible that certain neutrophil defects are specific features of T1D and not a general glucose-dependent defect. Future studies should look more carefully into neutrophil functions at different stages of diabetes.
2. Neutrophils in Type 2 Diabetes (T2D)
Type 2 diabetes mellitus (T2D) is a chronic disease characterized by an elevated concentration of glucose in blood as a result of limited insulin secretion and/or insulin resistance. As a consequence, in T2D, the metabolism of carbohydrates, lipids, and proteins is dysregulated [38].
T2D is associated with obesity-induced chronic systemic inflammation [39][40]. As described above, neutrophil infiltration into adipose tissues contributes to the development of insulin resistance. In mice fed a high-fat diet, neutrophils in the adipose tissue release NE which degrades IRS1, resulting in impaired insulin signaling [41] (Figure 2).
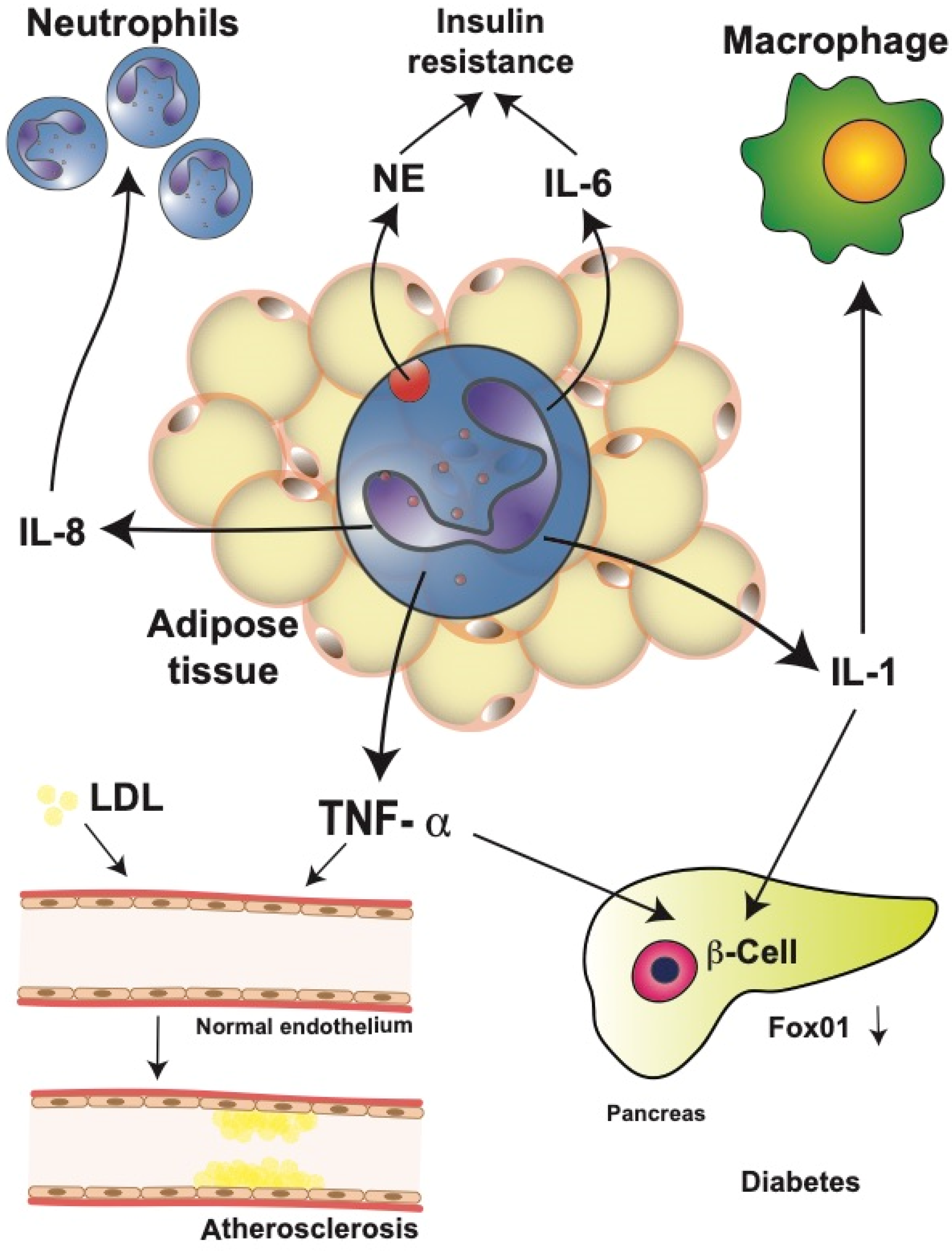
Figure 2. Neutrophils in obesity-related complications. Neutrophils in obese adipose tissue release large amounts of interleukin (IL)-1β, IL-6, IL-8, and tumor necrosis factor alpha (TNF-α). These cytokines have important systemic effects leading to obesity-related complications. IL-8 recruits more neutrophils into the adipose tissue, creating an amplification cycle. Neutrophil elastase (NE) and IL-6 contribute to the development of insulin resistance by impairing insulin signaling. IL-1β is an important activator of macrophages in multiple parts of the body. Furthermore, IL-1β, together with TNF-α in pancreatic islets, induces β cell dedifferentiation by downregulating transcription factor Fox01, which regulates β cell proliferation. Together, these events may result in type 2 diabetes mellitus. In addition, TNF-α can alter the adhesion function of endothelial cells by inducing increased low-density lipoprotein (LDL) uptake. These changes have been associated with the progression of atherosclerosis.
Therefore, genetically NE-deficient mice showed reduced adipose tissue inflammation and increased glucose tolerance, including better insulin sensitivity [41][42]. Furthermore, activated neutrophils from diabetic patients released more IL-1, IL-6, IL-8, and TNF-α than neutrophils from healthy individuals [43], leading to the increased level of circulating inflammatory cytokines. These elevated cytokines may then impact multiple organs in the body. One cytokine that has been repeatedly implicated in T2D is TNF-α [44][45]. Neutrophils from T2D patients secrete higher amounts of IL-6 and TNF-α in response to lipopolysaccharide (LPS) stimulation, resulting in insulin resistance, which then increases the blood glucose concentration [43]. Similarly, in the serum of obese patients with cardiovascular disease, larger TNF-α concentrations have been reported [46].
TNF-α has also been implicated in other mechanisms that contribute to the development of T2D, for example, altered function of endothelial cells [47][48]. Changes in the expression of adhesion molecules by vascular endothelial cells are observed in patients with T2D, and these changes seem to occur even before the onset of T2D [49]. Altered adhesion function of endothelial cells has also been associated with the progression of atherosclerosis [50]. TNF-α seems to be responsible for these alterations by inducing an increased low-density lipoprotein uptake in vascular endothelial cells [51] (Figure 2). This process can then promote atherosclerosis and extend inflammation [51]. Consequently, genetically TNF-α-deficient mice show less endothelial cell dysfunction in diabetes animal models [48][52]. In addition, TNF-α has been linked to β cell dysfunction and insulin resistance. TNF-α and IL-1 induced β cell dedifferentiation in cultured human and mouse pancreatic islets by downregulating transcription factor Fox01, which regulates β cell proliferation [53][54] (Figure 2).
The elevated levels of circulating inflammatory cytokines found in diabetic patients and animals also have important effects on neutrophil function. Neutrophils of diabetic individuals display lower phagocytic activity [33], lower production of ROS [37], and lower chemotactic capacity [55] than neutrophils from healthy control individuals. Some of these functions (migration and bacteria killing) also seem to be compromised in hyperglycemia, and can be induced in vitro upon exposure of neutrophils to serum from diabetic patients [56]. Finally, recent experiments showed that neutrophils can release microvesicles, which are involved in cell–cell communication. The neutrophil microvesicles concentration increased in the mice fed a high-fat diet. These microvesicles also accumulate in certain regions of arteries and promote vascular inflammation and atherosclerosis. In vitro, neutrophil microvesicles promoted inflammatory gene expression by endothelial cells [57]. Together, these reports suggest that neutrophils actively contribute to maintaining systemic inflammation and originating some pathological consequences found in T2D.
This entry is adapted from the peer-reviewed paper 10.3390/cells11121883
References
- Petersmann, A.; Müller-Wieland, D.; Müller, U.A.; Landgraf, R.; Nauck, M.; Freckmann, G.; Heinemann, L.; Schleicher, E. Definition, classification and diagnosis of diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 2019, 127, S1–S7.
- World Health Organization. Diabetes (Fact Sheet N°312). Available online: https://web.archive.org/web/20130826174444/http://www.who.int/mediacentre/factsheets/fs312/en/ (accessed on 25 January 2022).
- Demir, S.; Nawroth, P.P.; Herzig, S.; Ekim Üstünel, B. Emerging targets in type 2 diabetes and diabetic complications. Adv. Sci. 2021, 8, e2100275.
- Lotfy, M.; Adeghate, J.; Kalasz, H.; Singh, J.; Adeghate, E. Chronic complications of diabetes mellitus: A mini review. Curr. Diabetes Rev. 2017, 13, 3–10.
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55.
- Bollyky, J.B.; Xu, P.; Butte, A.J.; Wilson, D.M.; Beam, C.A.; Greenbaum, C.J.; Type 1 Diabetes TrialNet Study Group. Heterogeneity in recent-onset type 1 diabetes—A clinical trial perspective. Diabetes/Metab. Res. Rev. 2015, 31, 588–594.
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the non obese diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 2016, 66, 76–88.
- Chen, Y.G.; Mathews, C.E.; Driver, J.P. The role of NOD mice in type 1 diabetes research: Lessons from the past and recommendations for the future. Front. Endocrinol. 2018, 9, 51.
- Miao, D.; Yu, L.; Eisenbarth, G.S. Role of autoantibodies in type 1 diabetes. Front. Biosci. 2007, 12, 1889–1898.
- Kawasaki, E. Type 1 diabetes and autoimmunity. Clin. Pediatr. Endocrinol. 2014, 23, 99–105.
- Diana, J.; Simoni, Y.; Furio, L.; Beaudoin, L.; Agerberth, B.; Barrat, F.; Lehuen, A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 2013, 19, 65–73.
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013, 62, 2072–2077.
- Harsunen, M.H.; Puff, R.; D’Orlando, O.; Giannopoulou, E.; Lachmann, L.; Beyerlein, A.; von Meyer, A.; Ziegler, A.G. Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes. Horm. Metab. Res. 2013, 45, 467–470.
- Salami, F.; Lee, H.S.; Freyhult, E.; Elding Larsson, H.; Lernmark, Å.; Törn, C.; TEDDY Study Group. Reduction in white blood cell, neutrophil, and red blood cell counts related to sex, HLA, and islet autoantibodies in Swedish TEDDY children at increased risk for type 1 diabetes. Diabetes 2018, 67, 2329–2336.
- Vecchio, F.; Lo Buono, N.; Stabilini, A.; Nigi, L.; Dufort, M.J.; Geyer, S.; Rancoita, P.M.; Cugnata, F.; Mandelli, A.; Valle, A.; et al. Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight 2018, 3, e122146.
- Huang, J.; Xiao, Y.; Zheng, P.; Zhou, W.; Wang, Y.; Huang, G.; Xu, A.; Zhou, Z. Distinct neutrophil counts and functions in newly diagnosed type 1 diabetes, latent autoimmune diabetes in adults, and type 2 diabetes. Diabetes/Metab. Res. Rev. 2019, 35, e3064.
- Klocperk, A.; Petruzelkova, L.; Pavlikova, M.; Rataj, M.; Kayserova, J.; Pruhova, S.; Kolouskova, S.; Sklenarova, J.; Parackova, Z.; Sediva, A.; et al. Changes in innate and adaptive immunity over the first year after the onset of type 1 diabetes. Acta Diabetol. 2020, 57, 297–307.
- Qin, J.; Fu, S.; Speake, C.; Greenbaum, C.J.; Odegard, J.M. NETosis-associated serum biomarkers are reduced in type 1 diabetes in association with neutrophil count. Clin. Exp. Immunol. 2016, 184, 318–322.
- Dufort, M.J.; Greenbaum, C.J.; Speake, C.; Linsley, P.S. Cell type-specific immune phenotypes predict loss of insulin secretion in new-onset type 1 diabetes. JCI Insight 2019, 4, e125556.
- Shu, L.; Zhong, L.; Xiao, Y.; Wu, X.; Liu, Y.; Jiang, X.; Tang, T.; Hoo, R.; Zhou, Z.; Xu, A. Neutrophil elastase triggers the development of autoimmune diabetes by exacerbating innate immune responses in pancreatic islets of non-obese diabetic mice. Clin. Sci. 2020, 134, 1679–1696.
- Diana, J.; Lehuen, A. Macrophages and β-cells are responsible for CXCR2-mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol. Med. 2014, 6, 1090–1104.
- Garciafigueroa, Y.; Phillips, B.E.; Engman, C.; Trucco, M.; Giannoukakis, N. Neutrophil-associated inflammatory changes in the pre-diabetic pancreas of early-age NOD mice. Front. Endocrinol. 2021, 12, 565981.
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396.
- Németh, T.; Mócsai, A. The role of neutrophils in autoimmune diseases. Immunol. Lett. 2012, 143, 9–19.
- Wang, Y.; Xiao, Y.; Zhong, L.; Ye, D.; Zhang, J.; Tu, Y.; Bornstein, S.R.; Zhou, Z.; Lam, K.S.; Xu, A. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with β-cell autoimmunity in patients with type 1 diabetes. Diabetes 2014, 63, 4239–4248.
- Njeim, R.; Azar, W.S.; Fares, A.H.; Azar, S.T.; Kfoury Kassouf, H.; Eid, A.A. NETosis contributes to the pathogenesis of diabetes and its complications. J. Mol. Endocrinol. 2020, 65, R65–R76.
- Sodré, F.M.C.; Bissenova, S.; Bruggeman, Y.; Tilvawala, R.; Cook, D.P.; Berthault, C.; Mondal, S.; Callebaut, A.; You, S.; Scharfmann, R.; et al. Peptidylarginine deiminase inhibition prevents diabetes development in NOD mice. Diabetes 2021, 70, 516–528.
- Liang, Y.; Wang, X.; He, D.; You, Q.; Zhang, T.; Dong, W.; Fei, J.; Xing, Y.; Wu, J. Ameliorating gut microenvironment through staphylococcal nuclease-mediated intestinal NETs degradation for prevention of type 1 diabetes in NOD mice. Life Sci. 2019, 221, 301–310.
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819.
- Parackova, Z.; Zentsova, I.; Vrabcova, P.; Klocperk, A.; Sumnik, Z.; Pruhova, S.; Petruzelkova, L.; Hasler, R.; Sediva, A. Neutrophil extracellular trap-induced dendritic cell activation leads to Th1 polarization in type 1 diabetes. Front. Immunol. 2020, 11, 661.
- Smulski, C.R.; Eibel, H. BAFF and BAFF-receptor in B cell selection and survival. Front. Immunol. 2018, 9, 2285.
- Costa, S.; Bevilacqua, D.; Cassatella, M.A.; Scapini, P. Recent advances on the crosstalk between neutrophils and B or T lymphocytes. Immunology 2019, 156, 23–32.
- Wilson, R.M.; Reeves, W.G. Neutrophil phagocytosis and killing in insulin-dependent diabetes. Clin. Exp. Immunol. 1986, 63, 478–484.
- Cutler, C.W.; Eke, P.; Arnold, R.R.; Van Dyke, T.E. Defective neutrophil function in an insulin-dependent diabetes mellitus patients. A case report. J. Periodontol. 1991, 62, 394–401.
- Marhoffer, W.; Stein, M.; Schleinkofer, L.; Federlin, K. Evidence of ex vivo and in vitro impaired neutrophil oxidative burst and phagocytic capacity in type 1 diabetes mellitus. Diabetes Res. Clin. Pract. 1993, 19, 183–188.
- Kjersem, H.; Hilsted, J.; Madsbad, S.; Wandall, J.H.; Johansen, K.S.; Borregaard, N. Polymorphonuclear leucocyte dysfunction during short term metabolic changes from normo- to hyperglycemia in type 1 (insulin dependent) diabetic patients. Infection 1988, 16, 215–221.
- Nielson, C.P.; Hindson, D.A. Inhibition of polymorphonuclear leukocyte respiratory burst by elevated glucose concentrations in vitro. Diabetes 1989, 38, 1031–1035.
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of type 2 diabetes—Global burden of disease and forecasted trends. J. Epidemiol. Glob. Health 2020, 10, 107–111.
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445.
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180.
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412.
- Mansuy-Aubert, V.; Zhou, Q.L.; Xie, X.; Gong, Z.; Huang, J.Y.; Khan, A.R.; Aubert, G.; Candelaria, K.; Thomas, S.; Shin, D.J.; et al. Imbalance between neutrophil elastase and its inhibitor α1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab. 2013, 17, 534–548.
- Hatanaka, E.; Monteagudo, P.T.; Marrocos, M.S.; Campa, A. Neutrophils and monocytes as potentially important sources of proinflammatory cytokines in diabetes. Clin. Exp. Immunol. 2006, 146, 443–447.
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91.
- Good, M.; Newell, F.M.; Haupt, L.M.; Whitehead, J.P.; Hutley, L.J.; Prins, J.B. TNF and TNF receptor expression and insulin sensitivity in human omental and subcutaneous adipose tissue–influence of BMI and adipose distribution. Diabetes Vasc. Dis. Res. 2006, 3, 26–33.
- Bilgic Gazioglu, S.; Akan, G.; Atalar, F.; Erten, G. PAI-1 and TNF-α profiles of adipose tissue in obese cardiovascular disease patients. Int. J. Clin. Exp. Pathol. 2015, 8, 15919–15925.
- Meigs, J.B.; Hu, F.B.; Rifai, N.; Manson, J.E. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 2004, 291, 1978–1986.
- Lee, J.; Lee, S.; Zhang, H.; Hill, M.A.; Zhang, C.; Park, Y. Interaction of IL-6 and TNF-α contributes to endothelial dysfunction in type 2 diabetic mouse hearts. PLoS ONE 2017, 12, e0187189.
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74.
- Ntaios, G.; Gatselis, N.K.; Makaritsis, K.; Dalekos, G.N. Adipokines as mediators of endothelial function and atherosclerosis. Atherosclerosis 2013, 227, 216–221.
- Zhang, Y.; Yang, X.; Bian, F.; Wu, P.; Xing, S.; Xu, G.; Li, W.; Chi, J.; Ouyang, C.; Zheng, T.; et al. TNF-α promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: Crosstalk between NF-κB and PPAR-γ. J. Mol. Cell. Cardiol. 2014, 72, 85–94.
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNF alpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344.
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The role of inflammation in β-cell dedifferentiation. Sci. Rep. 2017, 7, 6285.
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234.
- Fainsod-Levi, T.; Gershkovitz, M.; Völs, S.; Kumar, S.; Khawaled, S.; Sagiv, J.Y.; Sionov, R.V.; Grunewald, M.; Keshet, E.; Granot, Z. Hyperglycemia impairs neutrophil mobilization leading to enhanced metastatic seeding. Cell Rep. 2017, 21, 2384–2392.
- Jafar, N.; Edriss, H.; Nugent, K. The effect of short-term hyperglycemia on the innate immune system. Am. J. Med. Sci. 2016, 351, 201–211.
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; West, L.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat. Commun. 2020, 11, 214.
This entry is offline, you can click here to edit this entry!