Cancer and neurodegeneration share leadership as causes of morbidity and death worldwide. They can be thought as disease mechanisms at opposite ends: while in neurodegeneration, induction of inflammatory genes and suppression of cell-cycle genes are the prominent signals; the opposite happens in cancer. Fenretinide (all-trans-N-(4-hydroxyphenyl) retinamide, 4-HPR) is a synthetic derivative of all-trans-retinoic acid initially proposed in anticancer therapy for its antitumor effects combined with limited toxicity. Currently, it is also studied in many other diseases for its ability to influence several biological pathways and provide a broad spectrum of pharmacological effects.
- fenretinide
- anticancer drug
- nanomicellar formulations
- repositioning
- neuroprotection
- hormesis
1. Fenretinide in Cancer
1.1 Clinical and Preclinical Evaluation of Fenretinide in Cancer
2. Fenretinide in Neurological Diseases
2.1 Preclinical evaluation of fenretinide in neurological disease
Multiple Sclerosis
In the experimental allergic encephalomyelitis (EAE) mice, low doses (3 mg/kg/day) of oral fenretinide, very far from those required to produce cytotoxic effects in tumor cells [50], were able to reduce severity of EAE symptoms [51]. Experimental autoimmune encephalomyelitis (EAE), in which susceptible mice strains are immunized with myelin basic protein (MBP), is one of the common models for relapsing/remitting multiple sclerosis (MS), an (auto)immune driven neurological disease specifically affecting the central nervous system.Spinal Cord Injury
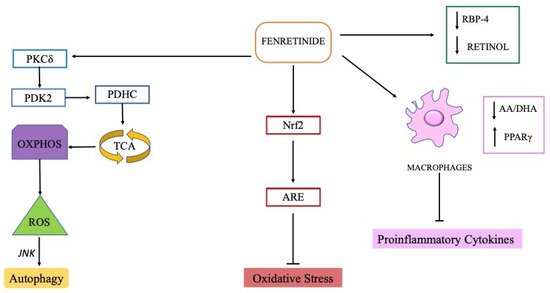
The omega-3 (DHA) and omega-6 (AA) polyunsaturated fatty acids (PUFA), major constituents of cell membranes, are the most susceptible to lipid peroxidation of all fatty acids. AA and the DHA act as potent signalling molecules that promote or dampen, respectively, molecular pathways involved in the inflammatory process [52].
Fenretinide, as already mentioned, down-regulates the activation of the ERK1/2 mitogen-activated protein (MAP) kinase pathway and the expression of inflammatory mediators via normalization of the AA/DHA balance [45][46]. Thus fenretinide can support the health of the central nervous system due to its ability to modulate fatty acid metabolism and lipid mediators.
This mechanism has been called into question in reducing expression of pro-inflammatory genes and decreasing oxidative stress in a mice model of spinal cord injury, in which fenretinide beneficially acted on motor deficit. Oral administration of low daily doses of fenretinide (5 mg/kg/die) modulated PUFA levels in plasma and in injured CNS tissue of mice, after spinal cord contusion. A significant improvement in locomotor recovery was also recorded [53].
Amyotrophic Lateral Sclerosis
The unpublished doctoral thesis of Skinner T [78], an author of the paper of Lopez-Vales et al. [53], describes a beneficial role of the same doses of fenretinide in a faster progressive mice model of amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease characterized by selective death of motor neurons causing progressive muscle atrophy and spasticity.
In spinal cord of ALS mice chronically treated (from presyptomatic stage of the disease) with fenretinide at low oral doses (5 mg/kg) Skinner observed increased DHA/AA ratio, decrease lipid peroxidation and gliosis (the hallmarks of neuroinflammation in ALS) and a significant reduction in the expression of TNF-α and iNOS proinflammatory mediators. These histopathologic findings well correlated with an improvement in motor functions and increased survival time in affected female mice.
Very recently we demonstrated that low doses (10 mg/kg) of a new nanomicellar fenretinide formulation, administered by intraperitoneal route, were able to extended survival of female, but non male, ALS mice even when administered after the onset of motor symptoms [54].
Alzheimer Disease
Retinoids can act upon multiple Alzheimer disease-associated targets, a neurodegenerative disease currently ranked as the most common cause of dementia among older adults [55].
Retinoid deficiency is greatest in late onset Alzheimer disease (LOAD) [56] which accounts for 90% of AD cases [57]. According, in a mouse model of AD it was shown that administration of all-trans retinoic acid recovered adult neurogenesis in the hippocampus through inhibition of microglial activation [58].
In a neuron-specific human BACE1 knock-in mice, an animal model of sporadic AD, fenretinide (0.04% w/w in diet, approximately corresponding to 40–50 mg/kg/die) has been shown to reduce adiposity gain and serum leptin, a key regulator hormone in the control of body weight, metabolism, and glucose homeostasis [59].
In the same animal model Plucińska and collaborators [60] demonstrated the ability of fenretinide (chronically administered with the diet) to decrease ER stress and neuroinflammation while boosting the expression of the sAPPα, the neuro-protective fragment of the amyloid precursor protein. In these mice fenretinide significantly prevented early signs of spatial memory deficits, restored the cerebral expression of full length APP and lowered the accumulation of toxic Aβ oligomers. However, fenretinide effects on AD phenotype appeared independent on its ability to modulate the systemic glucose homeostasis in BACE1 mice, suggesting that fenretinide may affect β-site cleavage of APP via an indirect mechanisms.
Depression
Lipopolysaccharide (LPS) administration is widely used to induce depressive-like behavior in rodents as LPS-treated mice mimick human depressive symptoms with an earlier “sickness behavior” phase which includes fever, anorexia, reduction of locomotion and a decrease in social interaction, followed by a several depressive-like behaviours [61].
LPS administration elevates the immobility time that reflects a measure of behavioral despair when placing a rodent in an uncomfortable situation from which escape is impossible; very interesting, intraperitoneal injections of fenretinide (20 and 40 mg/kg) reverted this depressive behaviour. Fenretinide also reduced BBB dysfunction in LPS-induced brain injury where promoted Nrf2-dependent antioxidative signalling [48].
Nrf2 pathway is naturally activated as an endogenous compensatory mechanism [62] and treatment with compounds able to cross the BBB and activate the Nrf2 pathway have been indicated as emerging therapeutic strategies against the oxidative stress damage in central nervous system, supporting the neuroptotective effect of fenretinide which exhibits these abilities [48]. Fenretinide can indirectly modulate antioxidant Nrf2 activity increasing β-carotene accumulation [96] that was shown to prevent neuronal loss in ROS-associated brain diseases also by Nrf2 increase [63][64]. In a phase 3 clinical trial (ClinicalTrials.gov Identifier: NCT00534898) fenretinide (5 mg/kg/die) was proposed as add-on to stable ongoing antipsychotic treatment in patients suffered from schizophrenia.
Retinopathies
Despite its peripheral location the retina, the neural portion of the eye, represents an extension of the central nervous system, with specialized immune responses similar to those of the brain and spinal cord.
Based on its ability to reduce circulating levels of retinol, fenretinide treatment has been proposed in geographic atrophy, an advanced form of age-related macular degeneration affecting the retina in which pathogenesis excessive accumulation of retinol-based toxins has been implicated. Unlike other organs, the uptake of retinol by the eye is largely dependent on delivery by RBP complex which can be efficiently modulated by fenretinide through a rapid elimination of the complex via urine.
In a 2-year placebo-controlled double-masked trial, 100 and 300 mg of fenretinide were orally administered every days in 246 geographic atrophy patients [65]. As expected, fenretinide administration induced a dose-dependent reversible reductions in serum RBP-retinol with visual disturbance and night blindness as the most common adverse effect in patients treated with the higher doses. More importantly, limiting retinol uptake from the serum into retinal pigment epithelium has been associated with a decrease in lesion growth rates, supporting the potential of fenretinide in the management of retinopathy.
3 Conclusion
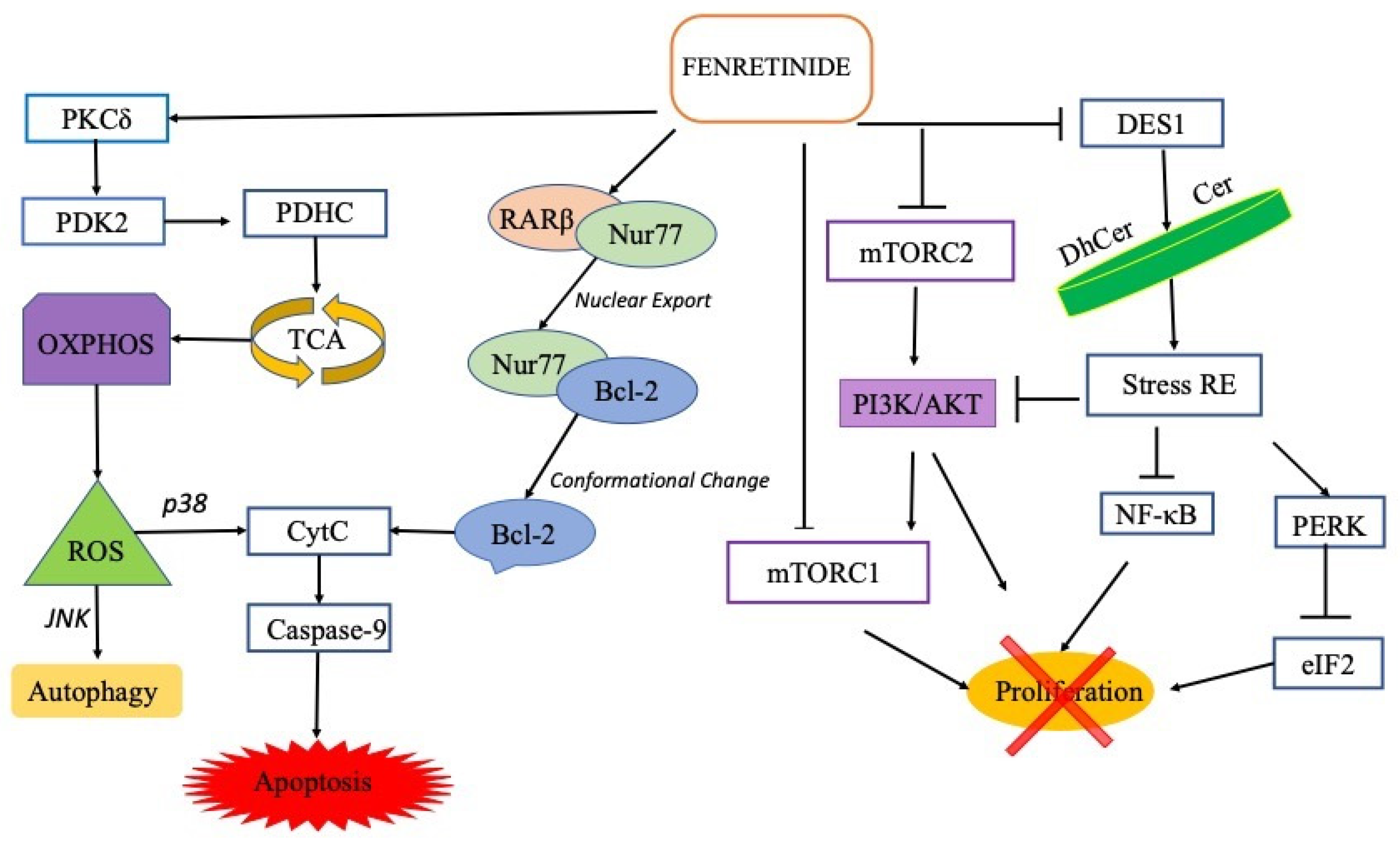
As already reported, high doses of fenretinide can activate acute responses leading to ROS increase and cell death [13], while, at subtoxic concentrations, fenretinide can stimulate adaptive stress responses [12]. Specifically, at lethal levels of oxidative stress, induced by high fenretinide concentrations, excessive oxidized DJ-1 (an oxidative stress response protein) induces p38 activation enabling the cells to commit to apoptosis. On the contrary, in the presence of mild oxidative stress, as provided by low fenretinide concentrations, mildly oxidized DJ-1 is recruited to inhibit the activity of ASK1 thus inhibiting apoptosis, maintaining cell viability by autophagy activation [13] and providing an overall antioxidant action [66].
Thus, fenretinide behaviour appears consistent with a hormetic-biphasic dose response. The hormetic behaviour of fenretinide, already reported for other carotenoids [67][68], is demonstrated by the different drug doses necessary to provide efficacy in tumours or neurological disease, respectively.
Therefore, the activity of fenretinide as pro-apoptotic/antitumor or pro-autophagic/adjuvant strictly depends on its concentration in the pathological site (tumor or CNS tissues). This concentration being related to the dose, the route of administration and the type of formulation.
The type of formulation has proved particularly important with fenretinide as this drug is characterized by extremely low aqueous solubility and hydrophobicity that strongly limit its availability in body fluids thus preventing its clinical use.
Among several formulations proposed, drug encapsulation and nanomicelle-like formulation (based on mixtures of phospholipids-liquid triglycerides with or without cyclodextrins, able to self-assemble in an aqueous environment) have demonstrated ability to increase fenretinide bioavailability at levels providing suitable plasma-tissues biodistribution and enhanced efficacy.
Recently, chemotherapeutic drugs such as kinase inhibitors, antimetabolites, alkylating agents and antibodies have been repurposed for neuroprotection, since it was demonstrated that some signalling pathways in neurodegeneration and cancer show peculiar sharing of genes and proteins that can become mutual therapeutic targets [69]. However, the major problems with the anticancer drugs use in neurodegenerative diseases are the presence of physical barriers in the brain, such as the blood brain barrier, drug resistance and neuronal toxicity.
The independence of fenretinide from Pgp and MRP [70] and the ability of fenretinide to cross the blood-brain barrier, due to its highly lipophilic nature, makes it a good candidate to treat different neurological diseases
The neurotoxicity of many anticancer drugs [71], that strongly restricts their repositioning in neurological disease, was not observed with fenretinide in long term administration studies as chemopreventive agent [72] and in many other studies as a chemotherapeutic drug [16][73]. Noteworthy, the ability of fenretinide to decrease retinol plasma levels by binding RBP4 can reduce the incidence of ATRA-induced neurotoxicity observed in paediatric oncological patients [74], thus representing a safer alternative to ATRA in cancer.
Therefore, considering the several preclinical evidences, the low toxicity profile, the independence from drug efflux transporters, the ability to penetrate the brain, the new available formulations and its hormetic behaviour, fenretinide can be regarded as potential drug candidate for selected neurological diseases, thus expanding its value beyond anticancer therapy
This entry is adapted from the peer-reviewed paper 10.3390/ijms23137426
References
- Simeone, A.-M.; Tari, A.M. How retinoids regulate breast cancer cell proliferation and apoptosis. Experientia 2004, 61, 1475–1484.
- Sani, B.P.; Shealy, Y.F.; Hill, D.L. N-(4-Hydroxyphenyl)retinamide: Interactions with retinoid-binding proteins/receptors. Carcinogenesis 1995, 16, 2531–2534.
- Yang, H.; Bushue, N.; Bu, P.; Wan, Y.-J.Y. Induction and intracellular localization of Nur77 dictate fenretinide-induced apoptosis of human liver cancer cells. Biochem. Pharmacol. 2010, 79, 948–954.
- Lin, B.; Kolluri, S.K.; Lin, F.; Liu, W.; Han, Y.-H.; Cao, X.; I Dawson, M.; Reed, J.C.; Zhang, X.-K. Conversion of Bcl-2 from Protector to Killer by Interaction with Nuclear Orphan Receptor Nur77/TR3. Cell 2004, 116, 527–540.
- Wu, L.; Chen, L. Characteristics of Nur77 and its ligands as potential anticancer compounds (Review). Mol. Med. Rep. 2018, 18, 4793–4801.
- Lovat, P.E.; Ranallic, M.; Petruzzellic, M.A.; Bernassolac, F.; Piacentinide, M.; Malcolm, A.J.; Pearson, A.D.; Melinoc, G.; Redfern, C.P. Effector Mechanisms of Fenretinide-Induced Apoptosis in Neuroblastoma. Exp. Cell Res. 2000, 260, 50–60.
- Lachkar, F.; Ferré, P.; Foufelle, F.; Papaioannou, A. Dihydroceramides: Their emerging physiological roles and functions in cancer and metabolic diseases. Am. J. Physiol. Metab. 2021, 320, E122–E130.
- Kraveka, J.M.; Li, L.; Szulc, Z.M.; Bielawski, J.; Ogretmen, B.; Hannun, Y.A.; Obeid, L.; Bielawska, A. Involvement of Dihydroceramide Desaturase in Cell Cycle Progression in Human Neuroblastoma Cells. J. Biol. Chem. 2007, 282, 16718–16728.
- Zheng, W.; Kollmeyer, J.; Symolon, H.; Momin, A.; Munter, E.; Wang, E.; Kelly, S.; Allegood, J.C.; Liu, Y.; Peng, Q.; et al. Ceramides and other bioactive sphingolipid backbones in health and disease: Lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1864–1884.
- Lai, W.-L.; Wong, N.-S. The PERK/eIF2α signaling pathway of Unfolded Protein Response is essential for N-(4-hydroxyphenyl)retinamide (4HPR)-induced cytotoxicity in cancer cells. Exp. Cell Res. 2008, 314, 1667–1682.
- Xie, H.; Zhu, F.; Huang, Z.; Lee, M.-H.; Kim, D.J.; Li, X.; Lim, D.Y.; Jung, S.K.; Kang, S.; Li, H.; et al. Identification of mammalian target of rapamycin as a direct target of fenretinide both in vitro and in vivo. Carcinogenesis 2012, 33, 1814–1821.
- Kim, Y.-K.; Hammerling, U. The mitochondrial PKCδ/retinol signal complex exerts real-time control on energy homeostasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158614.
- Cao, J.; Ying, M.; Xie, N.; Lin, G.; Dong, R.; Zhang, J.; Yan, H.; Yang, X.; He, Q.; Yang, B. The Oxidation States of DJ-1 Dictate the Cell Fate in Response to Oxidative Stress Triggered by 4-HPR: Autophagy or Apoptosis? Antioxid. Redox Signal. 2014, 21, 1443–1459.
- Tiwari, M.; Bajpai, V.K.; Sahasrabuddhe, A.A.; Kumar, A.; Sinha, R.A.; Behari, S.; Godbole, M.M. Inhibition of N-(4-hydroxyphenyl)retinamide-induced autophagy at a lower dose enhances cell death in malignant glioma cells. Carcinogenesis 2007, 29, 600–609.
- Cooper, J.P.; Reynolds, C.P.; Cho, H.; Kang, M.H. Clinical development of fenretinide as an antineoplastic drug: Pharmacology perspectives. Exp. Biol. Med. 2017, 242, 1178–1184.
- Garaventa, A.; Luksch, R.; Piccolo, M.S.L.; Cavadini, E.; Montaldo, P.G.; Pizzitola, M.R.; Boni, L.; Ponzoni, M.; DeCensi, A.; De Bernardi, B.; et al. Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma. Clin. Cancer Res. 2003, 9, 2032–2039.
- Formelli, F.; Cavadini, E.; Luksch, R.; Garaventa, A.; Villani, M.G.; Appierto, V.; Persiani, S. Pharmacokinetics of oral fenretinide in neuroblastoma patients: Indications for optimal dose and dosing schedule also with respect to the active metabolite 4-oxo-fenretinide. Cancer Chemother. Pharmacol. 2007, 62, 655–665.
- Villablanca, J.G.; Krailo, M.D.; Ames, M.M.; Reid, J.M.; Reaman, G.H.; Reynolds, C.P. Phase I trial of oral fenretinide in children with high-risk solid tumors: A report from the children’s oncology group (CCG 09709). J. Clin. Oncol. 2006, 24, 3423–3430.
- Vaishampayan, U.; Heilbrun, L.K.; Parchment, R.E.; Jain, V.; Zwiebel, J.; Boinpally, R.R.; LoRusso, P.; Hussain, M. Phase II trial of fenretinide in advanced renal carcinoma. Inves. New Drugs 2005, 23, 179–185.
- Schneider, B.J.; Worden, F.; Gadgeel, S.; Hodges, C.; Parchment, R.; Zwiebel, J.; Kraut, M.; Kalemkerian, G. Phase II study of fenretinide in patients with small cell lung cancer (SCLC) with progression after first- or second-line chemotherapy. J. Clin. Oncol. 2004, 22, 7299.
- Cheung, E.; Pinski, J.; Dorff, T.; Groshen, S.; Quinn, D.; Reynolds, C.P.; Maurer, B.J.; Lara, P.N.; Tsao-Wei, D.D.; Twardowski, P.; et al. Oral Fenretinide in Biochemically Recurrent Prostate Cancer: A California Cancer Consortium Phase II Trial. Clin. Genitourin. Cancer 2009, 7, 43–50.
- Moore, M.M.; Stockler, M.; Lim, R.; Mok, T.S.; Millward, M.; Boyer, M.J. A phase II study of fenretinide in patients with hormone refractory prostate cancer: A trial of the Cancer Therapeutics Research Group. Cancer Chemother. Pharmacol. 2010, 66, 845–850.
- Puduvalli, V.K.; Yung, W.A.; Hess, K.R.; Kuhn, J.G.; Groves, M.D.; Levin, V.A.; Zwiebel, J.; Chang, S.M.; Cloughesy, T.F.; Junck, L.; et al. Phase II Study of Fenretinide (NSC 374551) in Adults with Recurrent Malignant Gliomas: A North American Brain Tumor Consortium Study. J. Clin. Oncol. 2004, 22, 4282–4289.
- Colombo, N.; Formelli, F.; Cantù, M.G.; Parma, G.; Gasco, M.; Argusti, A.; Santinelli, A.; Montironi, R.; Cavadini, E.; Baglietto, L.; et al. A Phase I-II Preoperative Biomarker Trial of Fenretinide in Ascitic Ovarian Cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1914–1919.
- Reynolds, C.P.; Frgala, T.; Tsao-Wei, D.D.; Groshen, S.; Morgan, R.; McNamara, M.; Scudder, S.; Zwiebel, J.A.; Lenz, H.J.; Garcia, A.A. High plasma levels of fenretinide (4-HPR) were associated with improved outcome in a phase II study of recurrent ovarian cancer: A study by the California Cancer Consortium. J. Clin. Oncol. 2007, 25, 5555.
- Modiano, M.R.; Dalton, W.S.; Lippman, S.M.; Joffe, L.; Booth, A.R.; Meyskens, F.L., Jr. Phase II study of Fenretinide (N-retinamide) in advanced breast cancer and melanoma. Inves. New. Drugs 1990, 8, 317–319.
- Maurer, B.J.; Kang, M.H.; Villablanca, J.G.; Janeba, J.; Groshen, S.; Matthay, K.K.; Sondel, P.M.; Maris, J.M.; Jackson, H.A.; Goodarzian, F.; et al. Phase I trial of fenretinide delivered orally in a novel organized lipid complex in patients with relapsed/refractory neuroblastoma: A report from the new approaches to neuroblastoma therapy (NANT) consortium. Pediatr. Blood Cancer 2013, 60, 1801–1808.
- Thomas, J.S.; El-Khoueiry, A.B.; Maurer, B.J.; Groshen, S.; Pinski, J.K.; Cobos, E.; Gandara, D.R.; Lenz, H.J.; Kang, M.H.; Reynolds, C.P.; et al. A phase I study of intravenous fenretinide (4-HPR) for patients with malignant solid tumors. Cancer Chemother. Pharmacol. 2021, 87, 525–532.
- Orienti, I.; Salvati, V.; Sette, G.; Zucchetti, M.; Bongiorno-Borbone, L.; Peschiaroli, A.; Zolla, L.; Francescangeli, F.; Ferrari, M.; Matteo, C.; et al. A novel oral micellar fenretinide formulation with enhanced bioavailability and antitumour activity against multiple tumours from cancer stem cells. J. Exp. Clin. Cancer Res. 2019, 38, 373.
- Orienti, I.; Nguyen, F.; Guan, P.; Kolla, V.; Calonghi, N.; Farruggia, G.; Chorny, M.; Brodeur, G.M. A Novel Nanomicellar Combination of Fenretinide and Lenalidomide Shows Marked Antitumor Activity in a Neuroblastoma Xenograft Model. Drug Des. Dev. Ther. 2019, 13, 4305–4319.
- Orienti, I.; Farruggia, G.; Nguyen, F.; Guan, P.; Calonghi, N.; Kolla, V.; Chorny, M.; Brodeur, G.M. Nanomicellar Lenalidomide–Fenretinide Combination Suppresses Tumor Growth in an MYCN Amplified Neuroblastoma Tumor. Int. J. Nanomed. 2020, 15, 6873–6886.
- Upton, D.; Valvi, S.; Liu, J.; Yeung, N.; George, S.; Ung, C.; Khan, A.; Franshaw, L.; Ehteda, A.; Shen, H.; et al. DIPG-07. High throughput drug screening identifies potential new therapies for diffuse intrinsic pontine gliomas (Dipgs). Neuro Oncol. 2020, 22, iii288.
- Upton, D.; Valvi, S.; Liu, J.; Yeung, N.; George, S.; Ung, C.; Khan, A.; Franshaw, L.; Ehteda, A.; Shen, H.; et al. Rare-08. Potential new therapies for diffuse intrinsic pontine gliomas identified through high throughput drug screening. Neuro Oncol. 2021, 23, i42.
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74.
- Halliwell, B. Role of Free Radicals in the Neurodegenerative Diseases. Drugs Aging 2001, 18, 685–716.
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Protein Oxidation and Lipid Peroxidation in Brain of Subjects with Alzheimer’s Disease: Insights into Mechanism of Neurodegeneration from Redox Proteomics. Antioxid. Redox Signal. 2006, 8, 2021–2037.
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative Stress and Neurotoxicity. Chem. Res. Toxicol. 2007, 21, 172–188.
- Owens, T.; Babcock, A.A.; Millward, J.M.; Toft-Hansen, H. Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Res. Rev. 2005, 48, 178–184.
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114.
- Bassani, B.; Bartolini, D.; Pagani, A.; Principi, E.; Zollo, M.; Noonan, D.M.; Albini, A.; Bruno, A. Fenretinide (4-HPR) Targets Caspase-9, ERK 1/2 and the Wnt3a/β-Catenin Pathway in Medulloblastoma Cells and Medulloblastoma Cell Spheroids. PLoS ONE 2016, 11, e0154111.
- Guilbault, C.; Wojewodka, G.; Saeed, Z.; Hajduch, M.; Matouk, E.; De Sanctis, J.B.; Radzioch, D. Cystic Fibrosis Fatty Acid Imbalance Is Linked to Ceramide Deficiency and Corrected by Fenretinide. Am. J. Respir. Cell Mol. Biol. 2009, 41, 100–106.
- Saeed, S.; Guilbault, C.; De Sanctis, J.; Henri, J.; Marion, D.; St-Arnaud, R.; Radzioch, D. Fenretinide prevents the development of osteoporosis in Cftr-KO mice. J. Cyst. Fibros. 2008, 7, 222–230.
- Garić, D.; Dumut, D.C.; Shah, J.; De Sanctis, J.; Radzioch, D. The role of essential fatty acids in cystic fibrosis and normalizing effect of fenretinide. Cell Mol. Life Sci. 2020, 77, 4255–4267.
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013.
- Lachance, C.; Wojewodka, G.; Skinner, T.A.A.; Guilbault, C.; De Sanctis, J.B.; Radzioch, D. Fenretinide Corrects the Imbalance between Omega-6 to Omega-3 Polyunsaturated Fatty Acids and Inhibits Macrophage Inflammatory Mediators via the ERK Pathway. PLoS ONE 2013, 8, e74875.
- Lachance, C.; Segura, M.; Dominguez-Punaro, M.C.; Wojewodka, G.; De Sanctis, J.B.; Radzioch, D.; Gottschalk, M. Deregulated Balance of Omega-6 and Omega-3 Polyunsaturated Fatty Acids following Infection by the Zoonotic Pathogen Streptococcus suis. Infect. Immun. 2014, 82, 1778–1785.
- Lin, C.-H.; Lee, S.-Y.; Zhang, C.-C.; Du, Y.-F.; Hung, H.-C.; Wu, H.-T.; Ou, H.-Y. Fenretinide inhibits macrophage inflammatory mediators and controls hypertension in spontaneously hypertensive rats via the peroxisome proliferator-activated receptor gamma pathway. Drug Des. Dev. Ther. 2016, 10, 3591–3597.
- Li, T.; Zheng, L.-N.; Han, X.-H. Fenretinide attenuates lipopolysaccharide (LPS)-induced blood-brain barrier (BBB) and depressive-like behavior in mice by targeting Nrf-2 signaling. Biomed. Pharmacother. 2020, 125, 109680.
- Barone, E.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic. Biol. Med. 2021, 176, 16–33.
- Asumendi, A.; Morales, M.C.; Alvarez, A.; Aréchaga, J.; Perezyarza, G. Implication of mitochondria-derived ROS and cardiolipin peroxidation in N-(4-hydroxyphenyl)retinamide-induced apoptosis. Br. J. Cancer 2002, 86, 1951–1956.
- Racke, M.K.; Burnett, D.; Pak, S.H.; Albert, P.S.; Cannella, B.; Raine, C.S.; McFarlin, D.E.; Scott, D.E. Retinoid treatment of experimental allergic encephalomyelitis IL-4 production correlates with improved disease course. J. Immunol. 1995, 154, 450–458.
- Nadjar, A.; Leyrolle, Q.; Joffre, C.; Laye, S. Bioactive lipids as new class of microglial modulators: When nutrition meets neuroimunology. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 79, 19–26.
- Lopez-Vales, R.; Redensek, A.; Skinner, T.A.A.; Rathore, K.I.; Ghasemlou, N.; Wojewodka, G.; DeSanctis, J.; Radzioch, D.; David, S. Fenretinide Promotes Functional Recovery and Tissue Protection after Spinal Cord Contusion Injury in Mice. J. Neurosci. 2010, 30, 3220–3226.
- Orienti, I.; Armida, M.; Dobrowolny, G.; Pepponi, R.; Sollazzini, G.; Pezzola, A.; Casola, I.; Musarò, A.; Popoli, P.; Potenza, R.L. Fenretinide Beneficial Effects on Amyotrophic Lateral Sclerosis-associated SOD1G93A Mutant Protein Toxicity: In Vitro and In Vivo Evidences. Neuroscience 2021, 473, 1–12.
- Beheshti, S. Chapter 35—All-Trans Retinoic Acid in Alzheimer’s Disease. In Diagnosis and Management in Dementia; Martin, C.R., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 559–572. ISBN 9780128158548.
- Das, B.C.; Dasgupta, S.; Ray, S.K. Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer’s disease. Neural. Regen. Res. 2019, 14, 1880–1892.
- Bihaqi, S.W.; Schumacher, A.; Maloney, B.; Lahiri, D.K.; Zawia, N.H. Do epigenetic pathways initiate late onset Alzheimer disease (LOAD): Towards a new paradigm. Curr. Alzheimer Res. 2012, 9, 574–588.
- Takamura, R.; Watamura, N.; Nikkuni, M.; Ohshima, T. All-trans retinoic acid improved impaired proliferation of neural stem cells and suppressed microglial activation in the hippocampus in an Alzheimer’s mouse model. J. Neurosci. Res. 2016, 95, 897–906.
- Dekeryte, R.; Hull, C.; Plucinska, K.; Khan, S.; Kamli-Salino, S.; Mody, N.; Morrice, N.; McLaughlin, C.; Gault, V.; Platt, B.; et al. Effects of Liraglutide and Fenretinide treatments on the diabetic phenotype of neuronal human BACE1 knock-in mice. Biochem. Pharmacol. 2019, 166, 222–230.
- Plucińska, K.; Mody, N.; Dekeryte, R.; Shearer, K.; Mcilroy, G.D.; Delibegovic, M.; Platt, B. High-fat diet exacerbates cognitive and metabolic abnormalities in neuronal BACE1 knock-in mice—partial prevention by Fenretinide. Nutr. Neurosci. 2020, 25, 719–736.
- O’Connor, J.C.; A Lawson, M.; André, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2008, 14, 511–522.
- Sun, Y.; Yang, T.; Leak, R.K.; Chen, J.; Zhang, F. Preventive and Protective Roles of Dietary Nrf2 Activators Against Central Nervous System Diseases. CNS Neurol. Disord. Drug Targets 2017, 16, 326–338.
- Chen, P.; Li, L.; Gao, Y.; Xie, Z.; Zhang, Y.; Pan, Z.; Tu, Y.; Wang, H.; Han, Q.; Hu, X.; et al. β-carotene provides neuro protection after experimental traumatic brain injury via the Nrf2–ARE pathway. J. Integr. Neurosci. 2019, 18, 153–161.
- Park, H.-A.; Hayden, M.M.; Bannerman, S.; Jansen, J.; Crowe-White, K.M. Anti-Apoptotic Effects of Carotenoids in Neurodegeneration. Molecules 2020, 25, 3453.
- Mata, N.L.; Lichter, J.B.; Vogel, R.; Han, Y.; Bui, T.V.; Singerman, L.J. Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina 2013, 33, 498–507.
- Takahashi, N. Antioxidant Properties of N-(4-Hydroxyphenyl)retinamide (Fenretinide). Biol. Pharm. Bull. 2000, 23, 222–225.
- Cho, S.; Chae, J.S.; Shin, H.; Shin, Y.; Song, H.; Kim, Y.; Yoo, B.C.; Roh, K.; Cho, S.; Kil, E.-J.; et al. Hormetic dose response to L-ascorbic acid as an anti-cancer drug in colorectal cancer cell lines according to SVCT-2 expression. Sci. Rep. 2018, 8, 11372.
- Shin, J.; Saini, R.K.; Oh, J.-W. Low Dose Astaxanthin Treatments Trigger the Hormesis of Human Astroglioma Cells by Up-Regulating the Cyclin-Dependent Kinase and Down-Regulated the Tumor Suppressor Protein P53. Biomedicines 2020, 8, 434.
- Advani, D.; Gupta, R.; Tripathi, R.; Sharma, S.; Ambasta, R.K.; Kumar, P. Protective role of anticancer drugs in neurodegenerative disorders: A drug repurposing approach. Neurochem. Int. 2020, 140, 104841.
- A Roundhill, E.; A Burchill, S. Detection and characterisation of multi-drug resistance protein 1 (MRP-1) in human mitochondria. Br. J. Cancer 2012, 106, 1224–1233.
- Pellacani, C.; Eleftheriou, G. Neurotoxicity of antineoplastic drugs: Mechanisms, susceptibility, and neuroprotective strategies. Adv. Med Sci. 2020, 65, 265–285.
- Rotmensz, N.; De Palo, G.; Formelli, F.; Costa, A.; Marubini, E.; Campa, T.; Crippa, A.; Danesini, G.; Grottaglie, M.D.; Di Mauro, M.; et al. Long-term tolerability of fenretinide (4-HPR) in breast cancer patients. Eur. J. Cancer Clin. Oncol. 1991, 27, 1127–1131.
- Sabichi, A.L.; Modiano, M.R.; Lee, J.J.; Peng, Y.M.; Xu, M.J.; Villar, H.; Dalton, W.S.; Lippman, S.M. Breast tissue accumulation of retinamides in a randomized short-term study of fenretinide. Clin. Cancer Res. 2003, 9, 2400–2405.
- Lanvers, C.; Reinhardt, D.; Dübbers, A.; Wagner-Bohn, A.; Creutzig, U.; Ritter, J.; Boos, J. Pharmacology of all-trans-retinoic acid in children with acute promyelocytic leukemia. Med. Pediatr. Oncol. 2003, 40, 293–301.