Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Three out of four subtypes of arrestin proteins expressed in mammals self-associate, each forming oligomers of a distinct kind. Monomers and oligomers have different subcellular localization and distinct biological functions.
- arrestin
- oligomerization
- signaling
- conformation
1. Arrestins: A Small Family with Many Functions
Mammals express only four arrestin subtypes [1]. Visual arrestin (a.k.a. S-antigen, 48 kDa protein, and rod arrestin; systematic name arrestin-1) is expressed in rod and cone photoreceptor cells in the retina, whereas arrestin-4 is expressed exclusively in cones at a much lower level than arrestin-1 [2]. Both quench G protein-mediated signaling of photopigments [2,3]. The first family member discovered, arrestin-1 [4], ensures rapid recovery in rods [5,6,7] by shutting down light-induced signaling of the prototypical G protein-coupled receptor (GPCR) rhodopsin [8,9,10] with sub-second kinetics in mammals [11]. The first non-visual arrestin subtype was also discovered as a protein that turns off G protein-mediated receptor signaling [12]. It was termed β-arrestin (systematic name, arrestin-2) because it preferred β2-adrenergic receptor over rhodopsin, in contrast to arrestin-1 with the opposite preference [12,13]. The second cloned non-visual arrestin subtype also quenched G protein-mediated signaling and preferred β2-adrenergic receptor over rhodopsin. Therefore, it was termed β-arrestin2, with the retroactive renaming of the first one, β-arrestin1 [14]. The same protein was cloned from human thyroid and named hTHY-ARRX [15]. As by that time it became clear that non-visual arrestins bind not only β2-adrenergic receptor, but many other GPCRs, systematic nomenclature was proposed, where the number after “arrestin” indicated the order of cloning without implying anything else, so that β-arrestin2 was termed arrestin-3 [16]. The cone photoreceptor-specific arrestin was cloned later and termed X-arrestin in one study [17] and cone arrestin in another [18]. As it was cloned last, its systematic name is arrestin-4. Below, we use systematic names of arrestin proteins.
In addition to precluding GPCR coupling to cognate G proteins, both non-visual subtypes were shown to be involved in various branches of cellular signaling (reviewed in [19,20]), interacting with >100 partners each [21], i.e., demonstrating a remarkable versatility for an average-sized ~45 kDa protein (reviewed in [22]). The overall structures of arrestin-1 [23,24,25], arrestin-2 [26,27], arrestin-3 [28], and arrestin-4 [29] monomers are remarkably similar. However, all arrestin subtypes, with the sole exception of cone-specific arrestin-4, oligomerize [30], and the oligomers they form are strikingly different.
2. Arrestin-1
Arrestin-1 crystallized in two labs under different conditions, forming virtually identical tetramers [23,24] (Figure 1A,B), which suggested that it might oligomerize in vivo. Indeed, it was found to exist in a monomer–dimer–tetramer equilibrium in solution [31]. In the crystal tetramer, many elements of arrestin-1 known at the time to be involved in receptor binding were shielded by sister protomers. Thus, an idea that oligomers are storage forms was proposed [31]. Subsequent studies using pulse EPR technique double electron–electron resonance (DEER) showed that the shape of the tetramer of arrestin-1 in solution is very different from that of the crystal tetramer: the four protomers form a closed diamond [32] (Figure 2). This model of the solution tetramer was confirmed by successful disruption of arrestin-1 self-association with mutations targeting predicted protomer–protomer interaction sites [33,34]. In the solution tetramer and both possible dimers forming it, the receptor-binding elements of the monomers were also shielded by sister protomers. Moreover, it was directly demonstrated that only monomeric arrestin-1 binds rhodopsin [35], supporting the idea that if rhodopsin binding is its only function, oligomers must serve some other purpose. The study using multi-angle light scattering (MALS) confirmed that bovine arrestin-1 oligomerizes in solution, forming dimers that associate into tetramers [30]. Measurements of dimerization and tetramerization constants suggested that in case of bovine arrestin-1, tetramer formation is a cooperative process [30]. Monomer–dimer–tetramer equilibrium in solution was found to be a common feature of bovine, mouse, and human arrestin-1, although dimerization and tetramerization constants in these species are quite different [33]. Rods express enormous amounts of arrestin-1 (intracellular concentrations were estimated at ~2 mM), which is the second most abundant protein in these photoreceptors after rhodopsin [36,37,38]. In fact, the level of arrestin-1 in rods is 4–5 orders of magnitude greater than usual levels of the non-visual subtypes in “normal” cells, including neurons, which is in most cases ~20–200 nM [39,40]. Comparison of oligomerization constants with calculated concentrations of mouse arrestin-1 in photoreceptors suggested that the bulk of arrestin-1 in cells must be oligomeric. Apparently, the majority of arrestin-1 molecules in rods exist as dimers or tetramers, depending on the species [33]. The most parsimonious explanation of this is that oligomers are storage forms for arrestin-1 when it is free and not bound to rhodopsin.
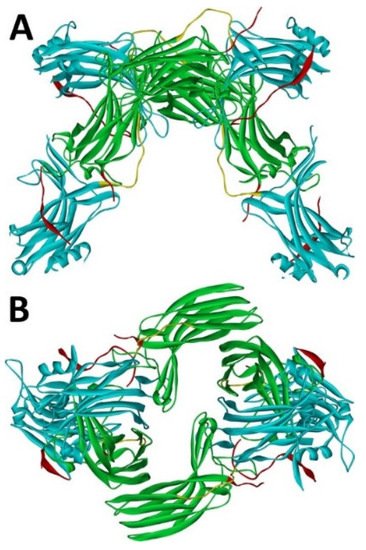
Figure 1. Crystal tetramer of bovine arrestin-1. (A), side view. (B), top view. PDB ID 1CF1 [24]. In each protomer arrestin-1 elements are colored, as follows: N-domain (residues 1–178), blue; inter-domain hinge (residues 179–190), yellow; C-domain (residues 191–356), green; C-tail (from residue 357; the gap reflects that part of the C-tail are invisible in crystal), red. Images were created in DS ViewerPro 6.0 (Dassault Systèmes, San Diego, CA, USA).
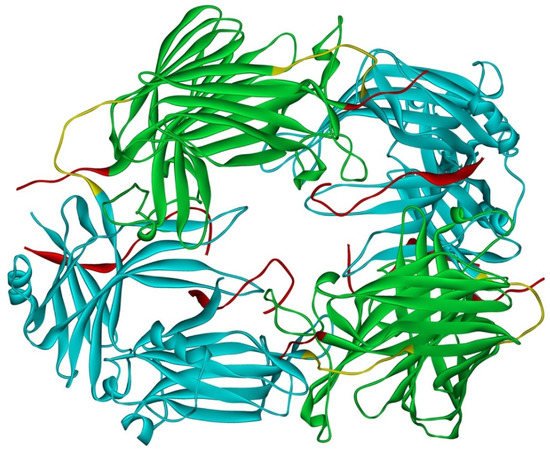
Figure 2. Solution tetramer of bovine arrestin-1. The image is based on the published model [32]. The color code is the same as in Figure 1. Image was created in DS ViewerPro 6.0 (Dassault Systèmes, San Diego, CA, USA).
However, it has not been clear why arrestin-1 needs to have these storage forms, i.e., why it could not be stored as a monomer. The first indication that the monomer might be harmful for rods came from in vivo experiments in mice using “enhanced” arrestin-1 mutant that binds unphosphorylated light-activated rhodopsin much better than wild type (WT) protein. Arrestin-1 binds with high affinity to active phosphorylated rhodopsin, shutting off the rhodopsin response to light and initiating a rapid recovery process (reviewed in [41]). Defects in rhodopsin phosphorylation or a lack of functional arrestin-1 in humans result in the loss of the rod function and night blindness [42,43]. Mice lacking rhodopsin kinase, which phosphorylates rhodopsin in rods upon light activation, or mice lacking arrestin-1 are effectively blind under normal light conditions, because rhodopsin signaling is not stopped, and therefore rods have no chance to recover [3,44]. We attempted to compensate for the lack of rhodopsin phosphorylation in rhodopsin kinase knockout mice by expressing a phosphorylation-independent arrestin-1 mutant in rods, which was expected to bind light-activated unphosphorylated rhodopsin and facilitate recovery. This compensational approach worked in principle, significantly reducing the recovery time [45,46]. However, the enhanced mutant used, which also happened to be partially oligomerization-deficient [34], when expressed either on the rhodopsin kinase or arrestin-1 knockout background, caused rapid degeneration of photoreceptor cells, the severity of which increased with its expression level [47]. Interestingly, consistent with the improved rhodopsin shutoff kinetics, the retinal degeneration caused by this mutant was light-independent. We have shown earlier that even very high supra-physiological levels of normally oligomerizing WT arrestin-1 were harmless for rods [36]. Furthermore, WT arrestin-1 co-expressed with the mutant dose-dependently protected, albeit partially, the rods from the mutant-induced degeneration [47]. These data suggested that the retinal degeneration caused by the arrestin-1 mutant might be due to an elevated concentration of a cytotoxic monomer. The protection afforded by WT arrestin-1 could then be easily explained by the known ability of oligomerization-competent WT arrestin-1 to draw partially oligomerization-deficient mutants into oligomers [47].
The oligomerization defect was not the only functional characteristic of the mutant different from that of WT arrestin-1. Another feature of the mutant, an enhanced interaction with clathrin adaptor AP2, was earlier shown to be harmful for the photoreceptor cells [48]. Thus, testing whether defective self-association per se is the culprit, required the replacement in vivo of WT arrestin-1 with an oligomerization-deficient mutant that otherwise has WT-like functional characteristics. An arrestin-1 with mutations that selectively impair oligomerization but do not affect rhodopsin binding was constructed and transgenically expressed in arrestin-1 knockout mice, replacing the WT protein [49]. A thorough analysis of these animals showed that non-oligomerizing protein normally quenches rhodopsin signaling [49], consistent with previous finding that arrestin-1 binds rhodopsin as a monomer [35]. However, arrestin-1 mutant with defective oligomerization and WT-like rhodopsin-binding characteristics caused progressive retinal degeneration, which was faster in lines with higher expression and did not depend on illumination at all, i.e., it proceeded at the same pace in dark-reared animals [49]. These data support the earlier conclusion that arrestin-1 monomer at high concentration is toxic for rod photoreceptors. Since rods do express arrestin-1 at very high levels [36,37,38], oligomerization might be required to prevent cytotoxicity. Calculations based on absolute arrestin-1 concentrations in mouse rods [36] and dimerization and tetramerization constants of mouse arrestin-1 [33] suggest that monomer concentration in WT rods is ~ 95 μM [49]. The data with mice expressing oligomerization-deficient arrestin-1 show that rods tolerate up to 500 μM of monomer, but higher concentrations are harmful [49]. Thus, available data suggest that monomeric and oligomeric forms of arrestin-1 are functionally different. The molecular mechanism whereby monomeric arrestin-1 exerts its cytotoxic effect still needs to be elucidated. It is also unknown whether arrestin-1 oligomers (dimers or tetramers) have specific functions in rods that the monomer cannot serve. These functions might be species-specific, as the predominant form of bovine and mouse arrestin-1 in the dark is likely a tetramer, whereas in humans dimers predominate [33].
In the dark, WT arrestin-1 is mostly localized to the rod inner segments and partially to the cell bodies and synaptic terminals, but in the light the bulk of it translocates to the rhodopsin-containing outer segments (OS) [50]. The arrestin-1 translocation has been shown to be a passive diffusion-driven process, with arrestin-1 moving along the gradients produced by its binding to the preferred partners in the light and dark [50,51]. When rhodopsin is activated by photons of light, it becomes rapidly phosphorylated by rhodopsin kinase. Then arrestin-1 monomer binds with high affinity to light-activated phosphorylated rhodopsin [35]. This shifts the monomer–dimer–tetramer equilibrium towards the monomer. As free arrestin-1 in the OS is depleted by its binding to rhodopsin, this process creates a concentration gradient driving the diffusion of arrestin-1 to the OS. In the dark, when rhodopsin is inactive, arrestin-1 is kept in other compartments via its binding to non-rhodopsin partners, most likely to microtubules particularly abundant in the inner segments [52]. Indeed, in the absence of higher affinity partners, such as light activated rhodopsin, arrestin-1 distribution resembles that of microtubules, and this is observed in the case of both WT arrestin-1 [50] and its oligomerization-deficient mutant [49].
Recently, an interesting model was proposed to explain the localization of arrestin-1 in the dark away from its “place of employment”, i.e., OS: oligomers were hypothesized to be too big to fit in the spaces between the rhodopsin-containing discs [53]. Indeed, the rod OS, where rhodopsin and the rest of the signal transduction machinery is localized, are filled with closely packed membranous discs housing rhodopsin with fairly narrow cytoplasmic spaces between them. This model predicts that in the dark, oligomerization-deficient arrestin-1 would be distributed in rod photoreceptors more evenly, with a significantly greater proportion localized to the OS. However, the subcellular distribution of an oligomerization-deficient mutant, which predominantly exists as a monomer, in both dark and light faithfully recapitulated that of the WT arrestin-1 [49], burying this beautifully simple model. The binding of arrestin-1 to the microtubules as a factor holding it in the dark in the inner segments and cell bodies remains the most likely explanation, particularly since both monomeric and oligomeric arrestin-1 bind microtubules well [35,54]. It is worth noting that while arrestin-1 has a microtubule-binding site overlapping with the rhodopsin footprint [54], which is shielded in the oligomeric forms [32], relatively low-affinity binding to microtubules appears to also be supported by the surfaces exposed in oligomers, as microtubule interaction, unlike rhodopsin binding, does not promote the dissociation of arrestin-1 oligomers into monomers [35].
3. Arrestin-2
Arrestin-2 crystallized as a dimer [26], which suggested that it also might oligomerize. Self-association of arrestin-2 in cells was described in 2005 [67]. Structural studies and extensive mutagenesis showed that arrestin-2 oligomerization involves two binding sites for an abundant cytosolic metabolite inositol-hexakisphosphate (IP6), one on the N- and the other on the C-domain, both on the concave (receptor-binding) side [68]. Importantly, the structure of arrestin-2 in crystals soaked with IP6 revealed long chains of arrestin-2, where the protomers interacted in an N-to-C-domain fashion, glued together by IP6 molecules between concave sides of the two domains of neighboring protomers [68] (Figure 3). This arrangement suggested that an oligomer is unlikely to bind GPCRs, as receptor-binding surfaces of arrestin-2 in it are shielded by neighboring protomers.
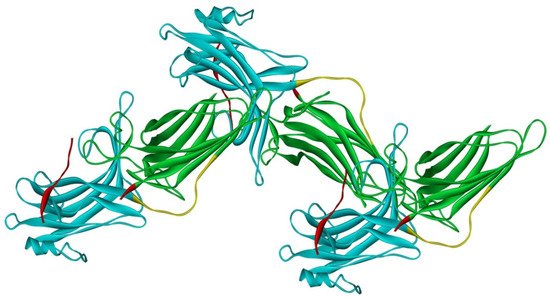
Figure 3. Crystal oligomer of bovine arrestin-2 in the presence of IP6. Based on PDB ID 1ZSH [68]. Arrestin-2 oligomer in the presence of IP6 appears to form the same “infinite” chains in solution [69], with each protomer in basal conformation, crystal structure of which was solved earlier (PDB ID 1G4M [26] and 1ZSH [27]). Arrestin-2 in crystal forms “infinite” chains with no apparent limit. Three molecules making IP6-mediated N-to-C contacts are shown. In each protomer arrestin-2 elements are colored, as follows: N-domain (residues 1–172), blue; inter-domain hinge (residues 173–184), yellow; C-domain (residues 185–352), green; C-tail (from residue 353), red (note the gap, as part of this element was not resolved in the crystal structure). Image was created in DS ViewerPro 6.0 (Dassault Systèmes, San Diego, CA, USA).
This entry is adapted from the peer-reviewed paper 10.3390/ijms23137253
This entry is offline, you can click here to edit this entry!