Glioblastomas are considered the most common and aggressive primary brain tumor in adults, with an average of 15 months’ survival rate. The treatment is surgery resection, followed by chemotherapy with temozolomide, and/or radiotherapy. Glioblastoma must have wild-type IDH gene and some characteristics, such as TERT promoter mutation, EGFR gene amplification, microvascular proliferation, among others. Glioblastomas have great heterogeneity at cellular and molecular levels, presenting distinct phenotypes and diversified molecular signatures in each tumor mass, making it difficult to define a specific therapeutic target. It is believed that the main responsibility for the emerge of these distinct patterns lies in subcellular populations of tumor stem cells, capable of tumor initiation and asymmetric division.
- glioblastoma
- brain tumor
- cancer stem cells
- molecular oncology
- chemoresistance
1. Introduction
1.1. Overview of Glioblastoma Classification, Mutations and Treatment
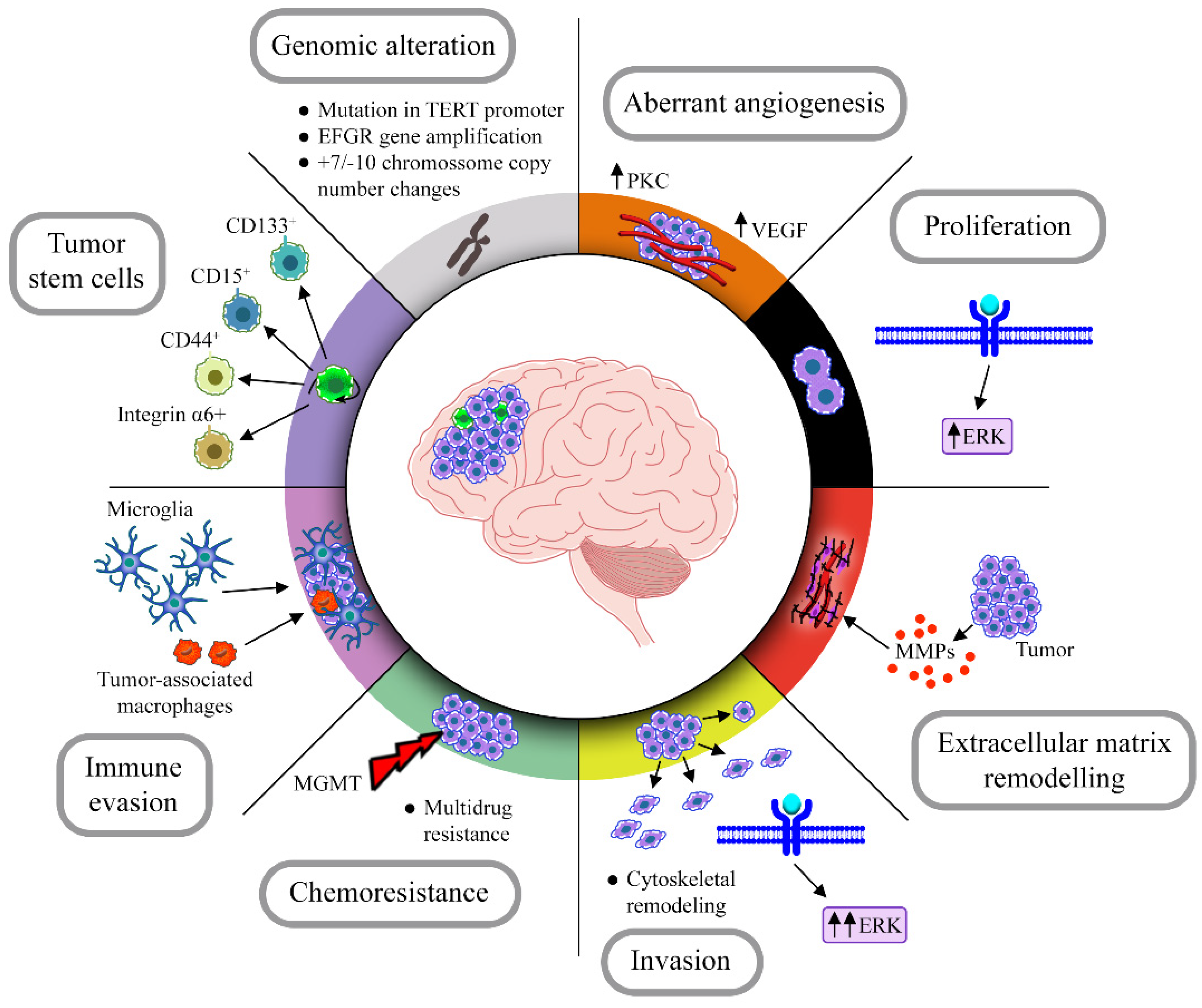
1.2. GBM Stem Cell Population: Implications in Tumor Resistance and Recurrence
2. IDH Mutation and Resistance Outcome
3. The Cytoskeleton as a Potential Therapeutic Target against Glioblastoma
3.1. Microtubules as Therapeutic Targets against Glioblastoma
3.2. Intermediate Filaments as Therapeutic Targets against Glioblastoma
3.3. Other Alternative Cytoskeletal Therapeutic Targets against Glioblastoma
This entry is adapted from the peer-reviewed paper 10.3390/cancers14133203
References
- Alexander, B.M.; Cloughesy, T.F. Adult glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409.
- Ohgaki, H.; Kleihues, P. Genetic Pathways to Primary and Secondary Glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453.
- Ostrom, Q.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncology 2014, 16, iv1–iv63.
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO Classification of Tumors of the Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225.
- Brat, D.J.; Van Meir, E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Investig. 2004, 84, 397–405.
- Schultz, S.; Pinsky, G.S.; Wu, N.C.; Chamberlain, M.C.; Rodrigo, A.S.; Martin, S.E. Fine needle aspiration diagnosis of extracranial glioblastoma multiforme: Case report and review of the literature. CytoJournal 2005, 2, 19.
- Linkous, A.G.; Yazlovitskaya, E.M. Angiogenesis in glioblastoma multiforme: Navigating the maze. Anti-Cancer Agents Med. Chem. 2011, 11, 712–718.
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417.
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251.
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820.
- Weinrich, S.L.; Pruzan, R.; Ma, L.; Ouellette, M.; Tesmer, V.M.; Holt, S.E.; Bodnar, A.G.; Lichtsteiner, S.; Kim, N.W.; Trager, J.B.; et al. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 1997, 17, 498–502.
- Bell, R.J.; Rube, H.T.; Xavier-Magalhães, A.; Costa, B.M.; Mancini, A.; Song, J.S.; Costello, J.F. Understanding TERT Promoter Mutations: A Common Path to Immortality. Mol. Cancer Res. 2016, 14, 315–323.
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026.
- Yuan, Y.; Qi, C.; Maling, G.; Xiang, W.; Yanhui, L.; Ruofei, L.; Yunhe, M.; Jiewen, L.; Qing, M. TERT mutation in glioma: Frequency, prognosis and risk. J. Clin. Neurosci. 2016, 26, 57–62.
- Inda, M.D.M.; Fan, X.; Muñoz, J.; Perot, C.; Fauvet, D.; Danglot, G.; Palacio, A.; Madero, P.; Zazpe, I.; Portillo, E.; et al. Chromosomal abnormalities in human glioblastomas: Gain in chromosome 7p correlating with loss in chromosome 10q. Mol. Carcinog. 2002, 36, 6–14.
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466.
- Wesolowski, J.; Rajdev, P.; Mukherji, S. Temozolomide (Temodar). Am. J. Neuroradiol. 2010, 31, 1383–1384.
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210.
- Cohen, M.H.; Johnson, J.R.; Pazdur, R. Food and Drug Administration Drug Approval Summary: Temozolomide Plus Radiation Therapy for the Treatment of Newly Diagnosed Glioblastoma Multiforme. Clin. Cancer Res. 2005, 11, 6767–6771.
- Abou-Antoun, T.; Hale, J.S.; Lathia, J.D.; Dombrowski, S.M. Brain Cancer Stem Cells in Adults and Children: Cell Biology and Therapeutic Implications. Neurotherapeutics 2017, 14, 372–384.
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828.
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Waschsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004, 23, 9392–9400.
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760.
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67.
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4, 440–452.
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martinez-Saez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668.
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; MacSwords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin Alpha 6 Regulates Glioblastoma Stem Cells. Cell Stem Cell 2010, 6, 421–432.
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-Mediated Dysfunction of the Bone Morphogenetic Protein Pathway Inhibits Differentiation of Glioblastoma-Initiating Cells. Cancer Cell 2008, 13, 69–80.
- Piccirillo, S.G.M.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; DiMeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765.
- Park, N.I.; Guilhamon, P.; Desai, K.; McAdam, R.F.; Langille, E.; O’Connor, M.; Lan, X.; Whetstone, H.; Coutinho, F.J.; Vanner, R.J.; et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell 2017, 21, 411.
- Sundar, S.J.; Hsieh, J.K.; Manjila, S.; Lathia, J.D.; Sloan, A. The role of cancer stem cells in glioblastoma. Neurosurg. Focus 2014, 37, E6.
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812.
- Narahara, K.; Kimura, S.; Kikkawa, K.; Takahashi, Y.; Wakita, Y.; Kasai, R.; Nagai, S.; Nishibayashi, Y.; Kimoto, H. Probable assignment of soluble isocitrate dehydrogenase (IDH1) to 2q33.3. Qual. Life Res. 1985, 71, 37–40.
- Geisbrecht, B.V.; Gould, S.J. The Human PICD Gene Encodes a Cytoplasmic and Peroxisomal NADP+-dependent Isocitrate Dehydrogenase. J. Biol. Chem. 1999, 274, 30527–30533.
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066.
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773.
- Reitman, Z.J.; Parsons, D.W.; Yan, H. IDH1 and IDH2: Not Your Typical Oncogenes. Cancer Cell 2010, 17, 215–216.
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1α. Science 2009, 324, 261–265.
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744.
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234.
- Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498.
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30.
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.-M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469.
- Mole, D.R.; Schlemminger, I.; McNeill, L.A.; Hewitson, K.S.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. 2-Oxoglutarate analogue inhibitors of hif prolyl hydroxylase. Bioorg. Med. Chem. Lett. 2003, 13, 2677–2680.
- Koivunen, P.; Lee, S.; Duncan, C.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488.
- Williams, K.; Christensen, J.; Helin, K. DNA methylation: TET proteins—Guardians of CpG islands? EMBO Rep. 2011, 13, 28–35.
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567.
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483.
- El-Habr, E.; Dubois, L.G.F.; Burel-Vandenbos, F.; Bogeas, A.; Lipecka, J.; Turchi, L.; Lejeune, F.-X.; Coehlo, P.L.C.; Yamaki, T.; Wittmann, B.M.; et al. A driver role for GABA metabolism in controlling stem and proliferative cell state through GHB production in glioma. Acta Neuropathol. 2016, 133, 645–660.
- Didiasova, M.; Banning, A.; Brennenstuhl, H.; Jung-Klawitter, S.; Cinquemani, C.; Opladen, T.; Tikkanen, R. Succinic Semialdehyde Dehydrogenase Deficiency: An Update. Cells 2020, 9, 477.
- Latini, A.; Scussiato, K.; Rosa, R.B.; Llesuy, S.; Belló-Klein, A.; Dutra-Filho, C.S.; Wajner, M. D-2-hydroxyglutaric acid induces oxidative stress in cerebral cortex of young rats. Eur. J. Neurosci. 2003, 17, 2017–2022.
- Struys, E.A.; Korman, S.H.; Salomons, G.S.; Darmin, P.S.; Achouri, Y.; Van Schaftingen, E.; Verhoeven, N.M.; Jakobs, C. Mutations in phenotypically mildD-2-hydroxyglutaric aciduria. Ann. Neurol. 2005, 58, 626–630.
- Struys, E.A.; Salomons, G.S.; Achouri, Y.; Van Schaftingen, E.; Grosso, S.; Craigen, W.J.; Verhoeven, N.M.; Jakobs, C. Mutations in the d-2-Hydroxyglutarate Dehydrogenase Gene Cause d-2-Hydroxyglutaric Aciduria. Am. J. Hum. Genet. 2005, 76, 358–360.
- Kranendijk, M.; Struys, E.A.; van Schaftingen, E.; Gibson, K.M.; Kanhai, W.A.; van der Knaap, M.S.; Amiel, J.; Buist, N.R.; Das, A.M.; de Klerk, J.B.; et al. IDH2 Mutations in Patients with d -2-Hydroxyglutaric Aciduria. Science 2010, 330, 336.
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516.
- Kim, E.S. Enasidenib: First Global Approval. Drugs 2017, 77, 1705–1711.
- Mellinghoff, I.K.; Penas-Prado, M.; Peters, K.B.; Burris, H.A.; Maher, E.A.; Janku, F.; Cote, G.M.; de la Fuente, M.I.; Clarke, J.L.; Ellingson, B.M.; et al. Vorasidenib, a Dual Inhibitor of Mutant IDH1/2, in Recurrent or Progressive Glioma; Results of a First-in-Human Phase I Trial. Clin. Cancer Res. 2021, 27, 4491–4499.
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468.
- Ramaekers, F.C.; Bosman, F.T. The cytoskeleton and disease. J. Pathol. 2004, 204, 351–354.
- Skalli, O.; Wilhelmsson, U.; Örndahl, C.; Fekete, B.; Malmgren, K.; Rydenhag, B.; Pekny, M. Astrocytoma grade IV (glioblastoma multiforme) displays 3 subtypes with unique expression profiles of intermediate filament proteins. Hum. Pathol. 2013, 44, 2081–2088.
- Katsetos, C.D.; Reginato, M.J.; Baas, P.W.; D’Agostino, L.; Legido, A.; Tuszyn’ski, J.A.; Dráberová, E.; Dráber, P. Emerging Microtubule Targets in Glioma Therapy. Semin. Pediatr. Neurol. 2015, 22, 49–72.
- Trendowski, M. Exploiting the cytoskeletal filaments of neoplastic cells to potentiate a novel therapeutic approach. Biochim. Biophys. Acta 2014, 1846, 599–616.
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803.
- Fellner, S.; Bauer, B.; Miller, D.S.; Schaffrik, M.; Fankhänel, M.; Spruß, T.; Bernhardt, G.; Graeff, C.; Färber, L.; Gschaidmeier, H.; et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J. Clin. Investig. 2002, 110, 1309–1318.
- Vellimana, A.K.; Recinos, V.R.; Hwang, L.; Fowers, K.D.; Li, K.W.; Zhang, Y.; Okonma, S.; Eberhart, C.G.; Brem, H.; Tyler, B.M. Combination of paclitaxel thermal gel depot with temozolomide and radiotherapy significantly prolongs survival in an experimental rodent glioma model. J. Neuro-Oncol. 2012, 111, 229–236.
- Elinzano, H.; Glantz, M.; Mrugala, M.; Kesari, S.; Piccioni, D.E.; Kim, L.; Pan, E.; Yunus, S.; Coyle, T.; Timothy, K.; et al. PPX and Concurrent Radiation for Newly Diagnosed Glioblastoma Without MGMT Methylation. Am. J. Clin. Oncol. 2018, 41, 159–162.
- Jeyapalan, S.; Boxerman, J.; Donahue, J.; Goldman, M.; Kinsella, T.; Dipetrillo, T.; Evans, D.; Elinzano, H.; Constantinou, M.; Stopa, E.; et al. Paclitaxel Poliglumex, Temozolomide, and Radiation for Newly Diagnosed High-grade Glioma. Am. J. Clin. Oncol. 2014, 37, 444–449.
- Régina, A.; Demeule, M.; Ché, C.; Lavallée, I.; Poirier, J.; Gabathuler, R.; Béliveau, R.; Castaigne, J.-P. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. J. Cereb. Blood Flow Metab. 2008, 155, 185–197.
- Silvani, A.; De Simone, I.; Fregoni, V.; Biagioli, E.; Marchioni, E.; Caroli, M.; Salmaggi, A.; Pace, A.; Torri, V.; Gaviani, P.; et al. Multicenter, single arm, phase II trial on the efficacy of ortataxel in recurrent glioblastoma. J. Neuro-Oncol. 2019, 142, 455–462.
- Manley, P.E.; Trippett, T.; Smith, A.A.; Macy, M.E.; Leary, S.E.; Boklan, J.; Cohen, K.J.; Goldman, S.; Kilburn, L.B.; Dhall, G.; et al. A phase 1/2 dose-finding, safety, and activity study of cabazitaxel in pediatric patients with refractory solid tumors including tumors of the central nervous system. Pediatr. Blood Cancer 2018, 65, e27217.
- Goldlust, S.; Nabors, L.B.; Duic, J.P.; Conrad, C.; Silberman, S.; Singer, S.; Farmer, G. At-24phase 1/2 trial of bevacizumab plus tpi 287, a novel brain penetrable anti-microtubule agent, for the treatment of recurrent glioblastoma. Neuro-Oncology 2014, 16, v13–v14.
- Oehler, C.; Frei, K.; Rushing, E.J.; McSheehy, P.M.; Weber, D.; Allegrini, P.R.; Weniger, D.; Lütolf, U.M.; Knuth, A.; Yonekawa, Y.; et al. Patupilone (Epothilone B) for Recurrent Glioblastoma: Clinical Outcome and Translational Analysis of a Single-Institution Phase I/II Trial. Oncology 2012, 83, 9152.
- Stupp, R.; Tosoni, A.; Bromberg, J.E.C.; Hau, P.; Campone, M.; Gijtenbeek, J.; Frenay, M.; Breimer, L.; Wiesinger, H.; Allgeier, A.; et al. Sagopilone (ZK-EPO, ZK 219477) for recurrent glioblastoma. A phase II multicenter trial by the European Organisation for Research and Treatment of Cancer (EORTC) Brain Tumor Group. Ann. Oncol. 2011, 22, 2144–2149.
- Peereboom, D.M.; Supko, J.G.; Carson, K.A.; Batchelor, T.; Phuphanich, S.; Lesser, G.; Mikkelsen, T.; Fisher, J.; Desideri, S.; He, X.; et al. A phase I/II trial and pharmacokinetic study of ixabepilone in adult patients with recurrent high-grade gliomas. J. Neuro-Oncology 2010, 100, 261–268.
- Brada, M.; Stenning, S.; Gabe, R.; Thompson, L.C.; Levy, D.; Rampling, R.; Erridge, S.; Saran, F.; Gattamaneni, R.; Hopkins, K.; et al. Temozolomide Versus Procarbazine, Lomustine, and Vincristine in Recurrent High-Grade Glioma. J. Clin. Oncol. 2010, 28, 4601–4608.
- Chamberlain, M.C.; Brain Tumor Investigational Consortium (BTIC); Grimm, S.; Phuphanich, S.; Recht, L.; Zhu, J.Z.; Kim, L.; Rosenfeld, S.; Fadul, C.E. A phase 2 trial of verubulin for recurrent glioblastoma: A prospective study by the brain tumor investigational consortium (BTIC). J. Neuro-Oncol. 2014, 118, 335–343.
- Kirby, S.; Gertler, S.Z.; Mason, W.; Watling, C.; Forsyth, P.; Aniagolu, J.; Stagg, R.; Wright, M.; Powers, J.; Eisenhauer, E.A. Phase 2 study of T138067-sodium in patients with malignant glioma: Trial of the National Cancer Institute of Canada Clinical Trials Group. Neuro-Oncology 2005, 7, 183–188.
- Kirkpatrick, J.; Desjardins, A.; Quinn, J.; Rich, J.; Vredenburgh, J.; Sathornsumetee, S.; Gururangan, S.; Sidor, C.; Friedman, H.; Reardon, D. Phase II open-label, safety, pharmacokinetic and efficacy study of 2-methoxyestradiol nanocrystal colloidal dispersion administered orally to patients with recurrent glioblastoma multiforme. J. Clin. Oncol. 2007, 25, 2065.
- Lickliter, J.; Fida, R.; Wheeler, H.; Kichenadasse, G.; Yang, Q.; Ganju, V.; Briggs, P. Carboplatin combined with the vascular-disrupting agent CYT997 for recurrent glioblastoma multiforme. J. Clin. Oncol. 2010, 28, e13591.
- Kolb, E.A.; Gorlick, R.; Keir, S.T.; Maris, J.M.; Kang, M.H.; Reynolds, C.P.; Lock, R.B.; Carol, H.; Wu, J.; Kurmasheva, R.T.; et al. Initial testing (stage 1) of BAL101553, a novel tubulin binding agent, by the pediatric preclinical testing program. Pediatr. Blood Cancer 2014, 62, 1106–1109.
- Bai, R.-Y.; Staedtke, V.; Aprhys, C.M.; Gallia, G.L.; Riggins, G.J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro-Oncology 2011, 13, 974–982.
- Zhao, J.; Zhang, L.; Dong, X.; Liu, L.; Huo, L.; Chen, H. High Expression of Vimentin is Associated With Progression and a Poor Outcome in Glioblastoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 337–344.
- Glassy, M.C.; Hagiwara, H. Summary analysis of the pre-clinical and clinical results of brain tumor patients treated with pritumumab. Hum. Antibodies 2009, 18, 127–137.
- Gutin, P.H.; Wong, E.T. Noninvasive Application of Alternating Electric Fields in Glioblastoma: A Fourth Cancer Treatment Modality. Am. Soc. Clin. Oncol. Educ. Book 2012, 32, 126–131.
- Kirson, E.D.; Gurvich, Z.; Schneiderman, R.; Dekel, E.; Itzhaki, A.; Wasserman, Y.; Schatzberger, R.; Palti, Y. Disruption of Cancer Cell Replication by Alternating Electric Fields. Cancer Res. 2004, 64, 3288–3295.
- Kesari, S.; Ram, Z. Tumor-treating fields plus chemotherapy versus chemotherapy alone for glioblastoma at first recurrence: A post hoc analysis of the EF-14 trial. CNS Oncol. 2017, 6, 185–193.
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma. JAMA J. Am. Med Assoc. 2017, 318, 2306–2316.
- Zhu, J.-J.; Demireva, P.; Kanner, A.A.; Pannullo, S.; Mehdorn, M.; Avgeropoulos, N.; Salmaggi, A.; Silvani, A.; Goldlust, S.; David, C.; et al. Health-related quality of life, cognitive screening, and functional status in a randomized phase III trial (EF-14) of tumor treating fields with temozolomide compared to temozolomide alone in newly diagnosed glioblastoma. J. Neuro-Oncol. 2017, 135, 545–552.
- Markel, G.; Sapir, Y.; Mandel, I.; Hakim, M.; Shaked, R.; Meilin, E.; McClanahan, T.; Loboda, A.; Hashmueli, S.; Ben Moshe, T. Inhibition of the novel immune checkpoint CEACAM1 to enhance anti-tumor immunological activity. J. Clin. Oncol. 2016, 34, 3044.
- Field, C.M.; Coughlin, M.; Doberstein, S.; Marty, T.; Sullivan, W. Characterization of anillin mutants reveals essential roles in septin localization and plasma membrane integrity. Development 2005, 132, 2849–2860.
- Spiliotis, E.T.; Kinoshita, M.; Nelson, W.J. A Mitotic Septin Scaffold Required for Mammalian Chromosome Congression and Segregation. Science 2005, 307, 1781–1785.
- Mittal, S.; Klinger, N.V.; Michelhaugh, S.K.; Barger, G.R.; Pannullo, S.C.; Juhász, C. Alternating electric tumor treating fields for treatment of glioblastoma: Rationale, preclinical, and clinical studies. J. Neurosurg. 2018, 128, 414–421.
- Mehta, M.; Wen, P.; Nishikawa, R.; Reardon, D.; Peters, K. Critical review of the addition of tumor treating fields (TTFields) to the existing standard of care for newly diagnosed glioblastoma patients. Crit. Rev. Oncol. 2017, 111, 60–65.
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010.
- Austefjord, M.W.; Gerdes, H.-H.; Wang, X. Tunneling nanotubes: Diversity in morphology and structure. Commun. Integr. Biol. 2014, 7, e27934.
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes—From electrical signals to organelle transfer. J. Cell Sci. 2012, 125, 1089–1098.
- Ariazi, J.; Benowitz, A.; De Biasi, V.; Boer, M.D.; Cherqui, S.; Cui, H.; Douillet, N.; Eugenin, E.A.; Favre, D.; Goodman, S.; et al. Tunneling Nanotubes and Gap Junctions–Their Role in Long-Range Intercellular Communication during Development, Health, and Disease Conditions. Front. Mol. Neurosci. 2017, 10, 333.
- Pontes, B.; Viana, N.B.; Campanati, L.; Farina, M.; Neto, V.M.; Nussenzveig, H.M. Structure and elastic properties of tunneling nanotubes. Eur. Biophys. J. 2007, 37, 121–129.
- Matias, D.; Dubois, L.G.; Pontes, B.; Rosário, L.; Ferrer, V.P.; Balça-Silva, J.; Fonseca, A.C.C.; Macharia, L.W.; Romão, L.; Spohr, T.C.L.D.S.E.; et al. GBM-Derived Wnt3a Induces M2-Like Phenotype in Microglial Cells Through Wnt/β-Catenin Signaling. Mol. Neurobiol. 2018, 56, 1517–1530.
- Menezes, A.; dos Reis, G.H.; Oliveira-Nunes, M.C.; Mariath, F.; Cabanel, M.; Pontes, B.; Castro, N.G.; de Brito, J.M.; Carneiro, K. Live Cell Imaging Supports a Key Role for Histone Deacetylase as a Molecular Target during Glioblastoma Malignancy Downgrade through Tumor Competence Modulation. J. Oncol. 2019, 2019, 9043675.
- Formicola, B.; D’Aloia, A.; Magro, R.D.; Stucchi, S.; Rigolio, R.; Ceriani, M.; Re, F. Differential Exchange of Multifunctional Liposomes Between Glioblastoma Cells and Healthy Astrocytes via Tunneling Nanotubes. Front. Bioeng. Biotechnol. 2019, 7, 403.
- Zhang, L.; Zhang, Y. Tunneling nanotubes between rat primary astrocytes and C6 glioma cells alter proliferation potential of glioma cells. Neurosci. Bull. 2015, 31, 371–378.
- Civita, P.; Leite, D.M.; Pilkington, G. Pre-Clinical Drug Testing in 2D and 3D Human In Vitro Models of Glioblastoma Incorporating Non-Neoplastic Astrocytes: Tunneling Nano Tubules and Mitochondrial Transfer Modulates Cell Behavior and Therapeutic Response. Int. J. Mol. Sci. 2019, 20, 6017.
- Pinto, G.; Saenz-De-Santa-Maria, I.; Chastagner, P.; Perthame, E.; Delmas, C.; Toulas, C.; Moyal-Jonathan-Cohen, E.; Brou, C.; Zurzolo, C. Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem. J. 2021, 478, 21–39.
- Valdebenito, S.; Audia, A.; Bhat, K.P.; Okafo, G.; Eugenin, E.A. Tunneling Nanotubes Mediate Adaptation of Glioblastoma Cells to Temozolomide and Ionizing Radiation Treatment. iScience 2020, 23, 101450.