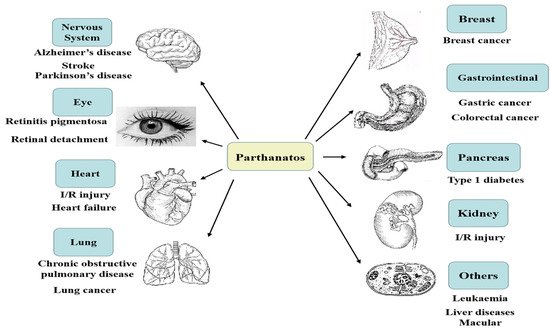
2. Hallmarks of Parthanatos
Cells undergoing parthanatos usually show necrosis-like and apoptosis-like morphological changes [
2]. These features include the loss of cell membrane integrity, cellular propidium iodide (PI) positive staining, and DNA fragmentations (15 kb to 50 kb) [
1,
11]. At the ultra-structural level, parthanatic cells usually exhibit mitochondrial abnormalities, such as the dissipation of inner transmembrane potential, nuclear shrinkage, and chromatin condensation [
1].
2.2.1. DNA Injury
DNA injury signaling is essential in maintaining genome integrity and cell fate. The main reasons for DNA injury are roughly divided into environmental effects (exogenous damage) and spontaneous injury (endogenous injury). Environmental factors often cover ultraviolet radiation (UVR), ionizing radiation (IR), alkylating agents, and metabolically activated compounds. Mistakes in DNA replication, base tautomerism, base deamination, and loss belong to spontaneous DNA damage. Sometimes chronic UVR is helpful to DNA repair response; most of the time, it results in unrepairable DNA damage [
12,
13,
14]. One publication emphasizes mitochondrial changes and DNA damage mediated by UVR in HaCaT cells, and the significance of PARP in these alterations [
15].
IR (α-, β-, γ-, X-ray), a valid and widely used measurement for cancer treatment, is able to control tumors affecting DNA damage response and repair (DRR) processes, which determine the fate of the tumor cells. IR-induced DNA double-strand breaks (DSBs) are the most lethal form of damage [
16].
Regarding alkylating agents, the primary mode of their action causes cytotoxic DNA damage. To resist alkylation-induced cell death or mutation, direct DNA damage reversal, base excision repair (BER), and mismatch repair (MMR) come into work. It is essential for a favorable response of the organism to alkylating agents to keep an appropriate balance of activity both within and between these pathways [
17]. Widespread as an environmental mutagen, N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) is a commonly used DNA-alkylating agent that potently initiates parthanatic cell death in cell-death research [
18].
ROS play a multifaceted and pleomorphic role in DNA damage response (DDR), such as mediating genotoxin-induced damage, DNA damage by oncogenic replication stress, sensing of DSBs, signal transduction within DDR, cell cycle progression, apoptosis, and DNA repair. It is necessary to distinguish the role between oxidative stress and ROS in DDR [
19,
20,
21].
PARP1 is crucial in maintaining genomic stability by facilitating DNA repair to ensure cell survival. It can regulate BER, single-stranded break (SSB), and DSB repair pathways. Meanwhile, PARP1 mediates parthanatos in response to severe DNA damage [
22].
2.2.2. NAD+ Depletion
Cellular energy depletion is caused by PARP1 over-activation through NAD
+ consumption [
23,
24]. NAD
+ is a co-factor in cellular metabolism, which is needed for generating ATP. Besides, NAD
+ resynthesis calls for about 2–4 molecules of ATP [
25]. Studies suggest that ATP decreases with the consumption of NAD
+, along with a decrease in cellular energy, resulting in cell death [
26,
27]. NAD
+ and energy are preserved when PARP1 inhibitors and the model of PARP1 deficiency are used [
28,
29]. However, the decrease in NAD
+ is not always associated with ATP decline in cerebral ischemia-reperfusion injury (CI/RI) models. A study points out that PARP1 KO mice reduce the infarct size in the CI/RI group, compared with the normal group, but the energy status does not change [
30].
2.2.3. Poly(ADP-Ribose) (PAR) Accumulation
The synthesis of PAR polymers depends on certain enzymes called PARPs [
31]. PARP1, a member of PARPs, is the most studied and makes the greatest contribution to synthesizing PAR polymers [
32]. When there is a toxic stimulus, DNA is damaged, PARP1 becomes hyper-activated and generates superfluous PAR polymer, followed by AIF nucleus translocation [
33]. PAR polymers activate a signal to modulate the downstream transcriptional process and the mechanism of DNA repair. PAR interacts with NONO, a novel PAR-binding protein, the binding modulates the physiological functions of protein [
34]. Subsequently, PAR has been studied to be directly toxic to neurons. AIF and PAR polymer-binding protein [
35,
36], a physical interaction between PAR and AIF, induces AIF release from the mitochondria and leads to the occurrence of parthanatos [
37,
38].
A few genes/proteins are considered biomarkers of parthanatos; for example, PARP1 is a therapeutic target in the stroke, trauma, I/R injury, and diabetes model [
23]. Cell parthanatos is suppressed in fibroblasts that are isolated from PARP1 KO mice [
39]. AIF is identified as a candidate, PAR-binding protein [
36,
37]. Genetic ablation of NAMPT or FK866 treatment sensitizes lymphocytes to MNNG-induced parthanatos, while over-expression of a catalytically active recombinant NAMPT protects NIH-3T3 cells from the toxicity of the same DNA alkylating agent [
40]. The result of AIF KO shows AIF as a cell death effector in PARP1 toxicity and parthanatos [
41]. KO PARG dysfunction sensitizes various cancer cells to chemotherapeutic agents and radiation [
42,
43]. Human osteosarcoma U2OS is exposed to hydrogen peroxide (H
2O
2) and is compared with the ADP-ribosylation (ADPr) pattern of control, ADP-ribosyl hydrolase 3 (ARH3) KO, HPF1 KO, and PARP1 KO cells. The data indicate that global ADPr in response to DNA damage requires ARH3, HPF1, and PARP1 [
44].
The activation of PARP1 promotes the transcription of pro-inflammatory genes and down-regulates multiple pathways of inflammation and tissue injury [
45]. The cells, which infect oligodendrocytes in human natural killer (NK) cells, are killed by virus-induced PARP1 and AIF translocation, rather than the immune system [
46]. For instance, the novel mechanism of immune escape in acute myeloid leukemia (AML) is uncovered: mature malignant cells in monocytic forms of leukemia have the capacity to produce ROS via NADPH oxidase and thus trigger parthanatos in adjacent antileukemic lymphocytes [
47]. In addition, the lymphocyte function is impaired because those malignant ROS-producing myeloid cells induce parthanatos in NK cells [
47,
48,
49]. NAD
+ metabolism makes a contribution to inflammation and immune responses [
50]. Human cytomegalovirus (HCMV) also restrains local immune responses through ROS-induced parthanatos [
14]. PARG is vital to tumor growth and metastasis of colon carcinoma and may be applied as a new target for treating colon carcinoma [
51].
3. Molecular Mechanisms of Parthanatos
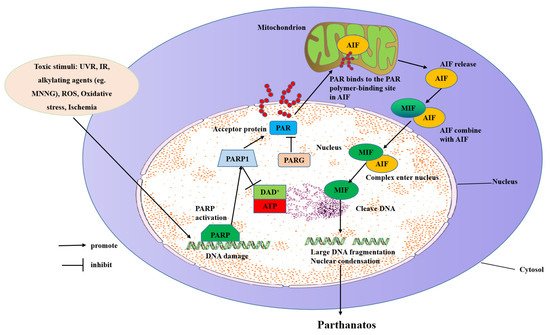
The molecular mechanisms of parthanatos include DNA damage, PARP1 hyper-activation, NAD+ and ATP depletion, and AIF translocation from the mitochondrial to the nucleus (Figure 2).
Figure 2. Molecular mechanisms of parthanatos. Toxic stimuli such as ROS, ischemia, alkylating agents (e.g., MNNG), IR, and UVR activate PARP-1 directly or indirectly through activation of NOS, which makes NO to induce ROS and the subsequent DNA damage. PARP-1 overactivation produces free PAR by PARG-mediated hydrolyzation, which serves as a death signal from the nucleus to mitochondria, inducing the release of AIF. AIF then translocates with MIF to the nucleus where it induces extensive fragmentation of DNA. This form of cell death is called “parthanatos”.
DNA damage is caused by toxic stimuli, such as H
2O
2 or hydroxyl radical, nitrosative stress from NO or peroxynitrite (ONOO
−), inflammation, ischemia or I/R, hypoxia, hypoglycemia, and DNA-alkylating agents [
25]. There are many DNA repair pathways, including BER, nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), and non-homologous end-joining (NHEJ). These pathways help to repair DNA damage throughout different stages of the cell cycle [
52].
PARP1 is a kind of enzyme that takes part in the process of DNA repair. It may not catalyze the process of DNA repair directly; however, the process of activating multiple DNA repair enzymes such as DNA topoisomerase, DNA helicase, and DNA ligase needs PARP1′s participation [
45]. PARP1 composes a DNA base-excision repair system through testing the crack and fracture of DNA strands and promoting its repair via PAR polymer synthesis. When DNA is mildly damaged, PARP1 is hundreds of times more active, and the enzyme uses NAD
+ to synthesize PAR polymer by consuming ATP. However, when there is serious DNA damage, PARP1 is over-activated and PAR polymers are generated and accumulated, which leads to phosphatidylserine externalization, mitochondrial membrane potential (MMP) dissipation, AIF nucleus translocation, massive DNA fragmentation, and chromatin condensation, and then parthanatos occurrence [
1,
2]. PAR also participates in DNA replication and repair; the cell, which is inhibited by PARG, shows an increased sensitivity to DNA-damaging agents [
10]. PARG is conducive to DSB and SSB repair, recovery from prolonged replication stress, and unusual replication structures in the S phase [
53].
PARP1, a highly expressed 116 kD eukaryotic nuclear protein, includes an N-terminal, a self-modifying domain, and a C-terminal catalytic domain. In response to DNA damage, histone PARylation factor 1 (HPF1), a required co-factor in PARP1/PARP2-mediated ADP-ribosylation of serine, directly binds to the catalytic domain of PARP1 via its C-terminal domain. The active site, which is formed by HPF1 and PARP1, activates the PARP1 enzyme [
54]. PARP1 combines with DNA primarily by the second zinc-finger domain [
45,
55,
56], it catalyzes NAD
+ and synthesizes PAR polymers adhered to various nuclear proteins, which significantly influences their function [
12].
PARP1 is vital in repairing DNA, leading to stability and transcription of genomics with normal physiological conditions. When DNA damage is slight, PARP1 is more reactive, resulting in ADP-ribosylation of PARP1 and its substrates, which then helps to recruit DNA repair effector proteins to repair DNA [
57]. Nevertheless, under immoderate genotoxic stress, PARP1 is hyper-activated and generates excessive PAR and stimulates AIF nucleus translocation, causing a sharp drop in NAD
+ storage after DNA breaks, which results in parthanatos. PARP1 is activated by DNA damage, which is caused by external stimuli. Furthermore, PARP1 is stimulated by the calcium (Ca
2+) signaling pathway [
9].
PARP1 hyper-activation is the first and critical step in the parthanatic cascade. The inhibition or deficiency of PARP1 is protective in models of the cell and rat/mouse injury paradigms, including retinal degeneration diseases, stroke, diabetes, I/R injury, neurodegenerative disease, and heart failure [
58]. PARP1 KO mouse models are hypersensitive to alkylating agents and IR, which is related to defective DNA repair [
59]. On the contrary, PARP1 KO mouse models are protected from LPS-induced shock, ischemic injury, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) excitotoxicity. Therefore, these genetic models emphasize a double function of PARP1 in the cell depending on the stimulation: promotion of DNA repair or mediation of pathological cascades of inflammation/excitotoxicity [
9]. These studies contribute to understanding the role of PARP1 in pathological contexts and related diseases. In preclinical zebrafish and human organotypic 3D skin models of psoriasis, PARP1 hyper-activation reacts to ROS-induced DNA damage: it is fueled by nicotinamide phosphoribosyltransferase-derived NAD
+ and mediates inflammation through parthanatos [
60]. In short, as a cell fate determinant, PARP1 has a significant influence in promoting DNA repair or restraining parthanatos. Now, many researchers have presented a number of potential treatments, of which the PARP1 enzyme has been regarded as a potential target intended to deal with related diseases. Targeting various diseases with chemical inhibitors of PARP1 provides therapeutic outcomes by reducing PARP1-mediated neuronal death. Many PARP1 inhibitors (oxaliplatin, PJ-34, 3-aminobenzamide, olaparib) have been studied in stroke, Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS), but have not been clinically evaluated [
61].
Nuclear DNA damage stimulation triggers the synthesis of poly ribose (ADP-ribose), next to the distribution of the PAR polymer to the cytoplasm and mitochondria, and then the release of AIF [13]. PAR inhibits the glycolysis process. ARH3 hydrolyzes protein-free PAR in the nucleus and cytoplasm, inhibits PAR transfer to the cytoplasm, and thus prevents parthanatos [62]. ARH3 has been shown to catalyze PAR-degradation in vitro [63,64]. PARG, the most well-characterized enzyme in humans for PAR hydrolysis, utilizes a macrodomain fold to bind ADP-ribosylation and specifically cleaves the ribose-ribose bonds between the subunits of the PAR chains. PARG needs the cooperation of PARP to repair DNA. Once the strand of PARP and DNA breaks, an enzyme is activated, causing PARP to shuttle and the chromatin to open. PARG enters the nucleus and moves to the PARP substrate to repair the broken DNA strand. With the increase in PARG and the decrease in PAR, chromatin recovers its original structure. PARG has been shown to play a vital role in various diseases [65].
NAD+ is a factor of ATP generation and its resynthesis needs many molecules of ATP, which its consumption results in ATP depletion and cellular energy downturn, which leads to parthanatos [9,28]. The inhibition of PARP1 by pharmacological inhibitors or genetic deletion could recover NAD+ levels [28]. Neurons that are stimulated with toxic show protection and accompanying energy conservation with PARP inhibitors. Besides, PARP1 KO mice of transient cerebral ischemia-induced damage present preserved NAD+ levels [29,66], supporting the suicide hypothesis of PARP1 activation causing a block in glycolysis. Besides, adding NAD+ to cells or over-expression of NAD+ avoids PARP1-dependent parthanatos, which suggests that the decreased NAD+ relevant to PARP1 hyper-activation causes cell demise [28].
A recent finding suggests that the mitochondrial pool of NAD
+ is associated with parthanatos because that parthanatos is saved by the preservation of NAD
+ in the mitochondria by adding NAD
+ biosynthetic enzyme Nampt or MPT inhibitor cyclosporine-A (CsA), and replenishing NAD
+ [
67,
68]. The decrease in NAD
+ after PARP1 hyper-activation reflects the level of NAD
+ in the whole cell. Therefore, the conclusions of these studies need to be re-examined as the research shows that mitochondrial NAD
+ is still at physiological levels after genotoxic stress, and even the nucleus and cytoplasmic NAD
+ are depleted [
68]. There are no reports yet to confirm that PARP1-dependent parthanatos entirely depends on mitochondrial NAD
+ depletion [
69]. Cyclosporine A may reduce parthanatos because it preserves mitochondrial NAD
+ and inhibits mitochondrial permeability transitions, which are themselves an essential participant in parthanatos [
67].
Researchers have questioned whether PARP1 hyper-activation was killed mainly by NAD
+ depletion. NAD
+ descent is not always associated with ATP decline in the I/R model [
70]. Moreover, the energy status does not change as the infarct size is reduced in PARP1 KO mice after MCAO compared with normal mice [
71]. In addition, Bax and calpain KO cells are protected from MNNG to the same degree as wild-type controls treated with the PARP1 inhibitor DPQ [
72]. Although the NAD
+ level is decreased in Bax and calpain KO cells, it is not in DPQ-treated cells. These results imply that PARP1 activation, which is caused by NAD
+ depletion, is insufficient to account for parthanatos. NAD
+ is expended by ADP-ribose transferases (ARTs) and many PARPs to generate an ADP-ribose protein modification and form PARP1 [
73]. cADP-ribose synthases generate and hydrolyze the Ca
2+-mobilizing second-messenger cADP-ribose from NAD
+ [
25,
69,
74]. There are two ways to promote the increase in NAD
+ levels: stimulating the synthesis of NAD
+ or inhibiting its excessive consumption [
25,
75,
76].
Mitochondria is a crucial element in the regulation of parthanatos. Its inter-membrane gap contains a number of proteins released through the outer membrane for taking part in parthanatos degradation. Mitochondria maintain the physiological integrity of cells by energy generation. However, once a cell is stimulated, the mitochondria become permeabilized, and AIF is released to irritate parthanatos. The change of outer membrane permeabilization (OMP) and permeability transition pore (PTP) will lead to the release of pro-apoptotic factors [77]. Bax, one of the pro-apoptotic members, is downstream of PARP1 and induces OMP needed for AIF release. Besides OMP, AIF cleavage by calpains requires AIF to be entirely released from the mitochondria [2].
According to the function, AIF is divided into three parts: an N-terminal part (binds to FAD), a central portion (which binds to NAD or NADH, which have oxidoreductase ability), and a C-terminal part (which is related to parthanatos) [
26]. The harlequin (Hq) mouse is a helpful model to research AIF-mediated parthanatos [
78]. It is noteworthy that AIF translocation is a vital step for parthanatos [
33,
79], so the understanding of this event is important in translating parthanatos research into therapy. In the nucleus, AIF-binding DNA is considered necessary even for parthanatos precipitation [
80]. AIF, a parthanatos effector, takes part in the process of generating mitochondrial energy. It is usually restricted to mitochondria but translocates to the nucleus when parthanatos happens. [
81,
82]. AIF translocation, a marker of parthanatos, usually occurs in conjunction with the detection of specific mitochondrial markers. AIF contains a PAR-binding motif, facilitating the direct association between PARP1 and AIF [
36], and takes part in PARP1 toxicity [
82]. When PARP1 is hyper-activated, excessive PAR escapes from the nucleus and binds to specific cytosolic or mitochondrial proteins [
35,
38]. PAR combines with these proteins and eventually causes AIF to be released from the mitochondria.
AIF’s structure is markedly regulated by binding NAD(P)H and forming a stable, long-lived charge-transfer complex (CTC). In the process of PARP1-initiated parthanatos, once the NAD
+ level is critically depleted, structural modulation of NADH regulates AIF release from mitochondria [
83,
84]. Excessive PAR triggers the mitochondrial release of AIF, which combines with macrophage MIF and carries MIF into the nucleus where the combination cleaves genomic DNA into large fragments. Chromatinolysis could be inhibited by depleting MIF and/or disrupting the AIF-MIF interaction then preventing MIF nuclease activity. The prevention of MIF nuclease activity should be a fascinating target for diseases.