Glucagon-like peptide-1 (GLP-1) is a human incretin hormone derived from the proglucagon molecule. GLP-1 receptor agonists are frequently used to treat type 2 diabetes mellitus and obesity. However, the hormone affects the liver, pancreas, brain, fat cells, heart, and gastrointestinal tract. The results showed that GLP-1 agonists can benefit defined off-medication motor scores in Parkinson’s Disease and improve emotional well-being. In Alzheimer’s disease, GLP-1 analogs can improve the brain’s glucose metabolism by improving glucose transport across the blood–brain barrier. In depression, the analogs can improve quality of life and depression scales. GLP-1 analogs can also have a role in treating chemical dependency, inhibiting dopaminergic release in the brain’s reward centers, decreasing withdrawal effects and relapses. These medications can also improve lipotoxicity by reducing visceral adiposity and decreasing liver fat deposition, reducing insulin resistance and the development of non-alcoholic fatty liver diseases. The adverse effects are primarily gastrointestinal. Therefore, GLP-1 analogs can benefit other conditions besides traditional diabetes and obesity uses.
- glucagon-like peptide-1
- non-alcoholic fatty liver disease
- Alzheimer’s disease
- depression
- diabetes
- obesity
- Parkinson’s disease
1. Introduction
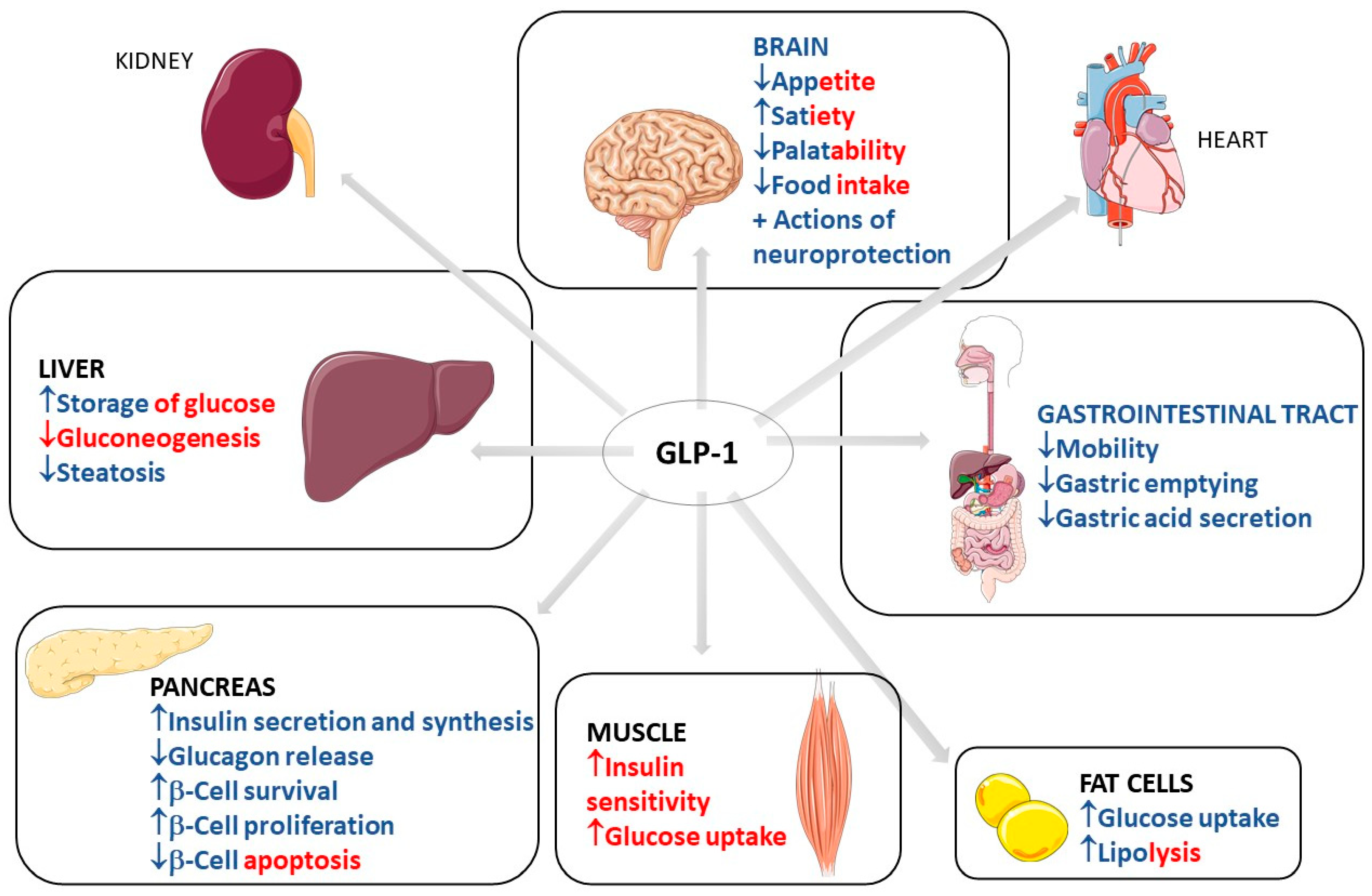
2. Use of GLP1
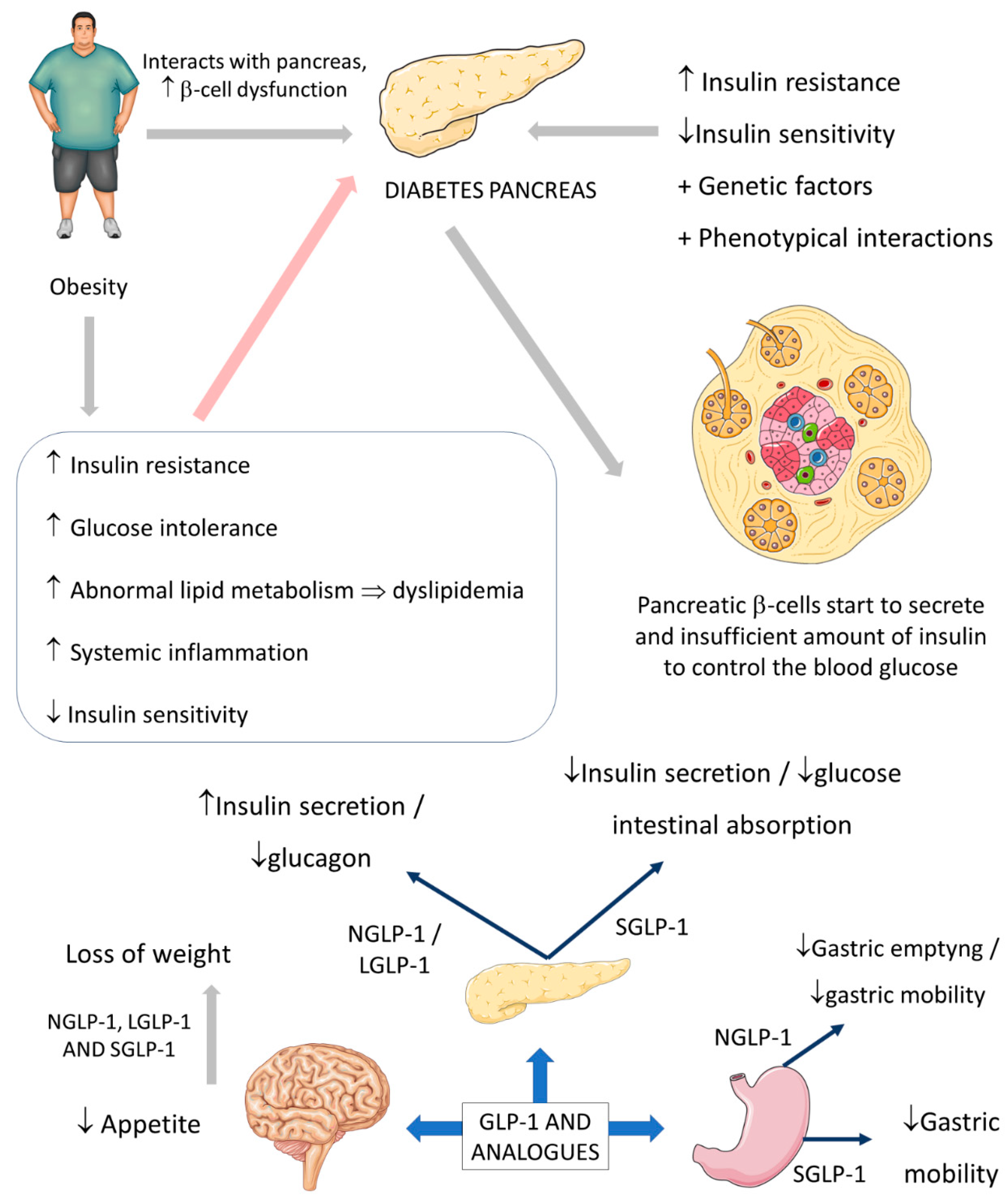
2.1. Common Use of GLP-1: Diabetes and Obesity
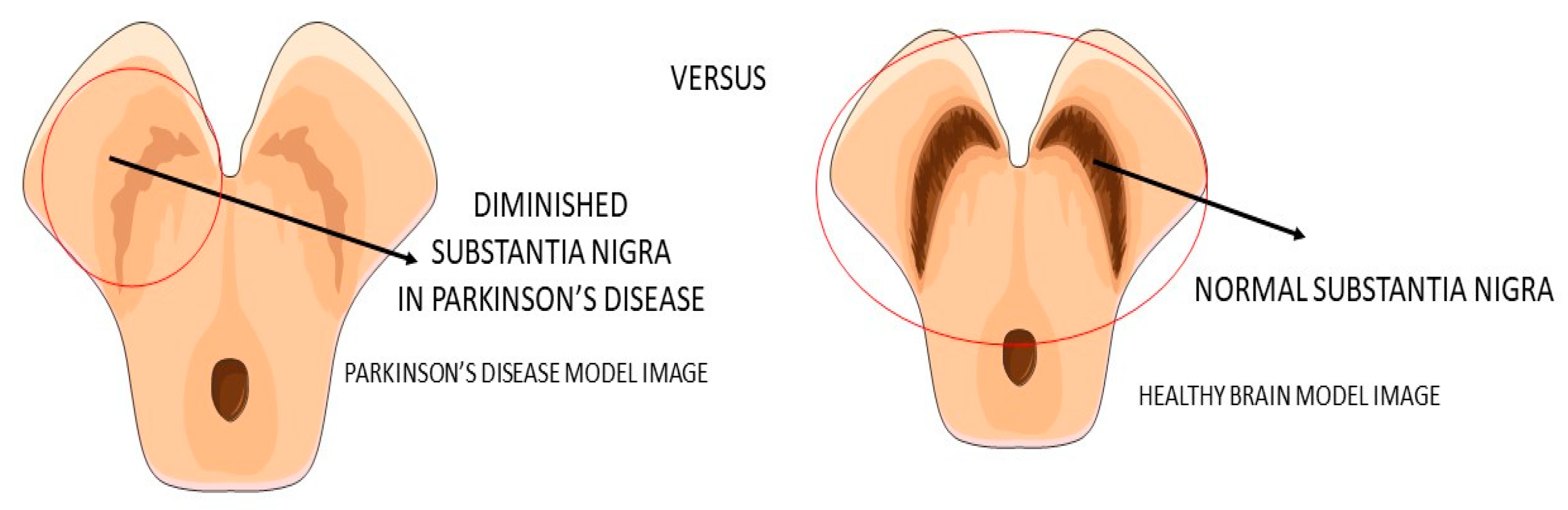
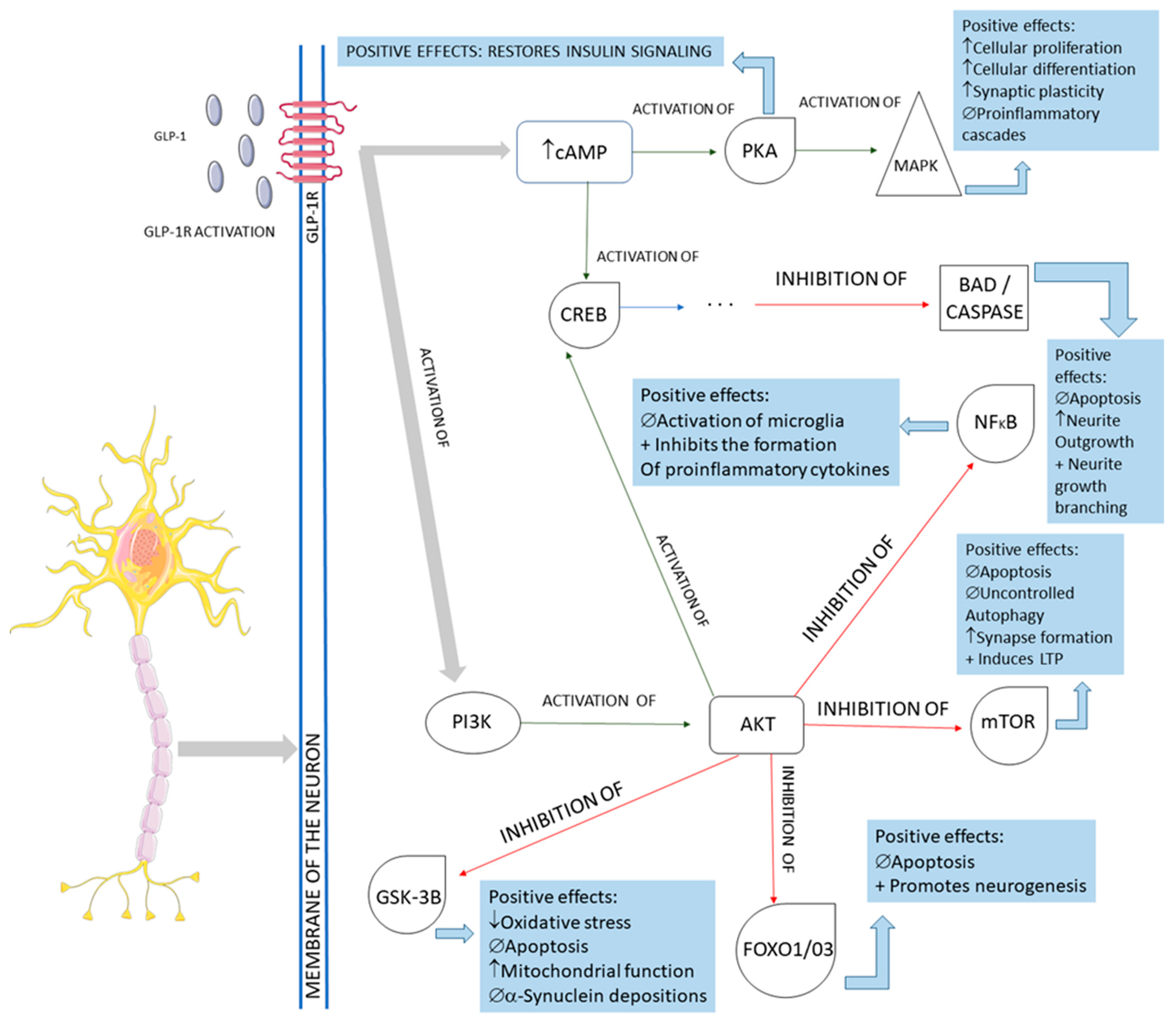
2.2. GLP-1 and Parkinson’s Disease
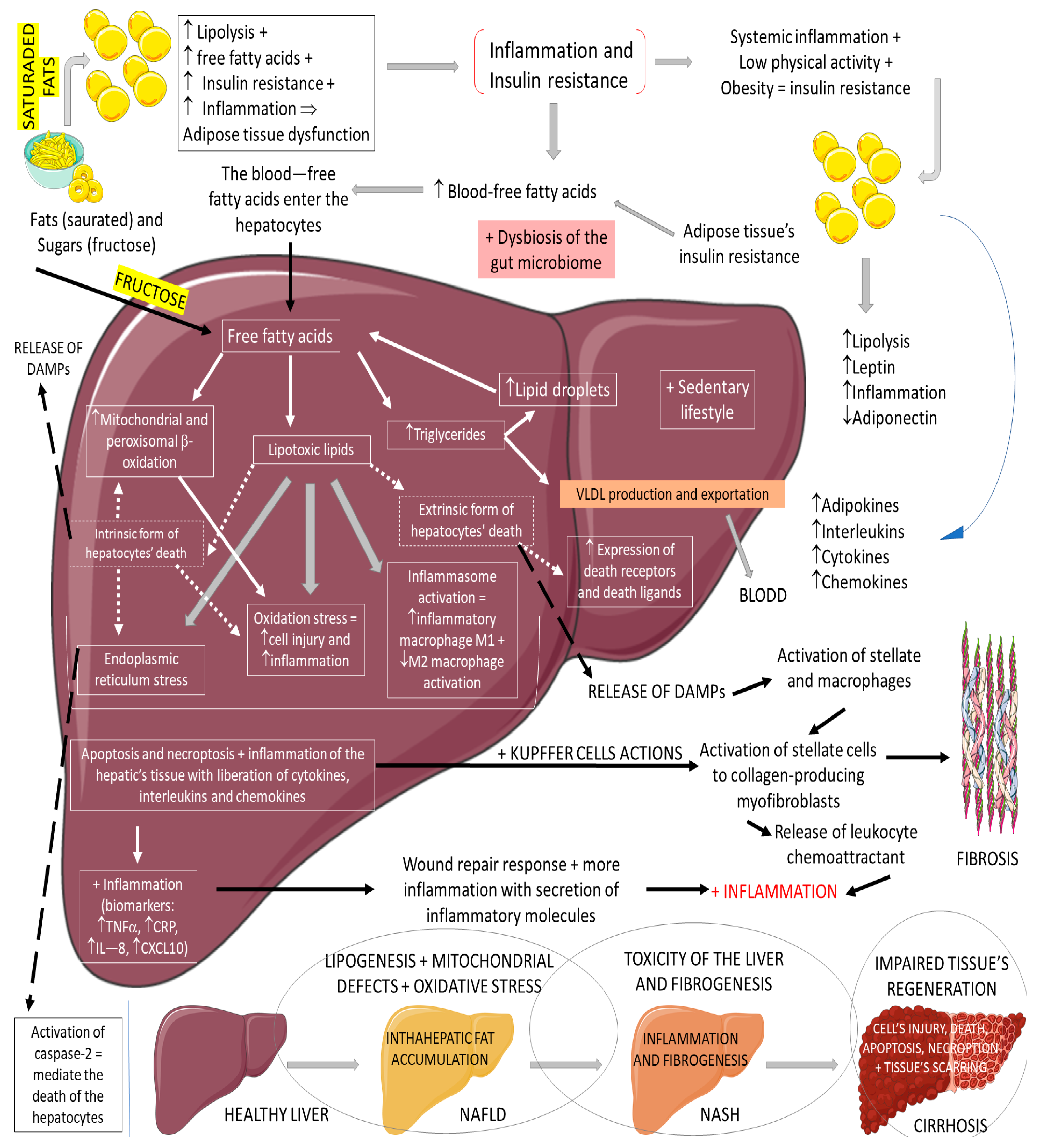
2.3. GLP-1, Non-Alcoholic Fatty Liver Disease, and Non-Alcoholic Steatohepatitis
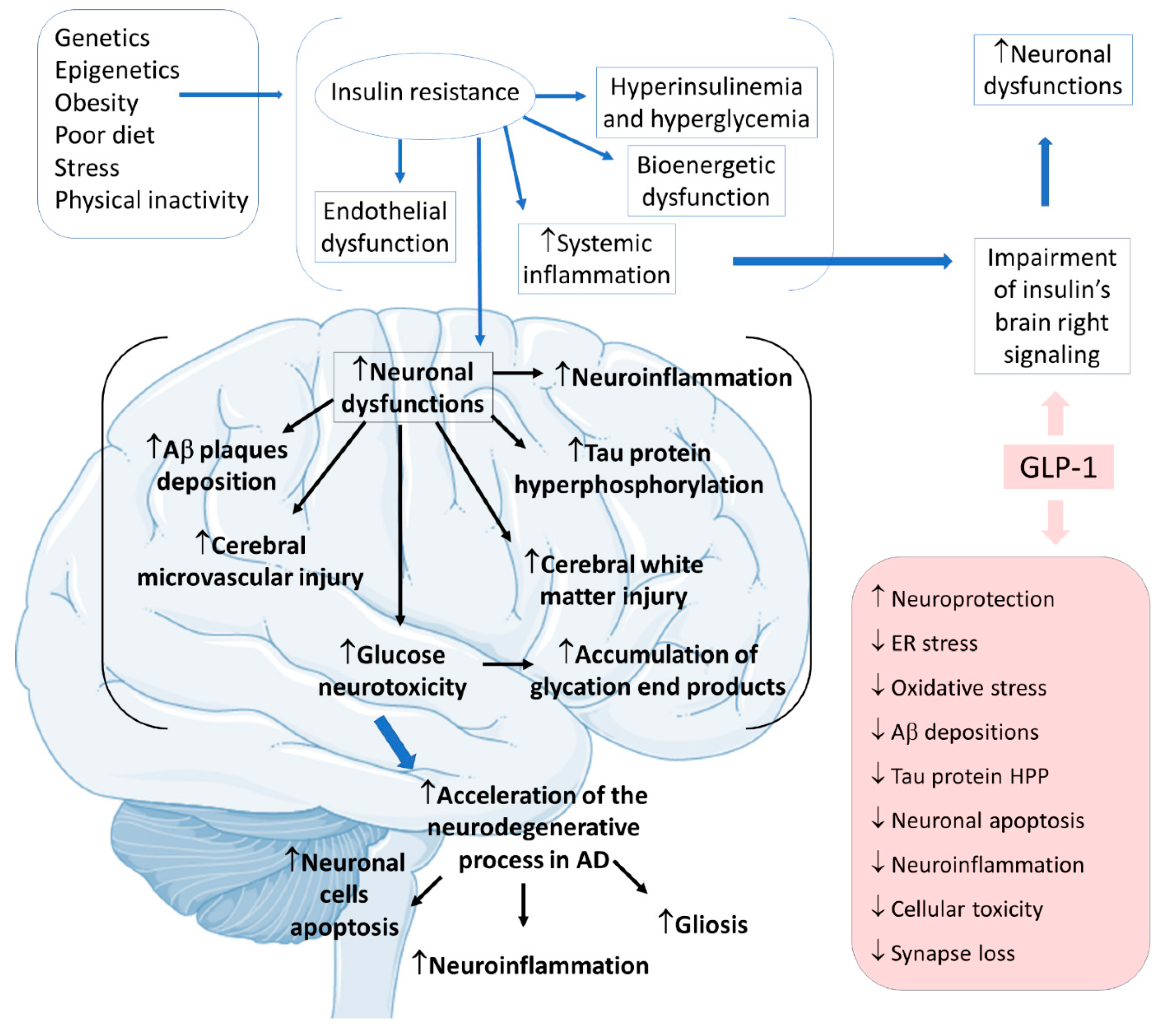
2.4. GLP-1 and Alzheimer’s Disease
2.5. GLP-1 and Depression
This entry is adapted from the peer-reviewed paper 10.3390/ijms23020739
References
- Smith, N.K.; Hackett, T.A.; Galli, A.; Flynn, C.R. GLP-1: Molecular mechanisms and outcomes of a complex signaling system. Neurochem. Int. 2019, 128, 94–105.
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130.
- Chia, C.W.; Egan, J.M. Incretins in obesity and diabetes. Ann. N. Y. Acad. Sci. 2020, 1461, 104–126.
- Erbil, D.; Eren, C.Y.; Demirel, C.; Küçüker, M.U.; Solaroğlu, I.; Eser, H.Y. GLP-1′s role in neuroprotection: A systematic review. Brain Inj. 2019, 33, 734–819.
- Gribble, F.M.; Reimann, F. Metabolic Messengers: Glucagon-like peptide 1. Nat. Metab. 2021, 3, 142–148.
- Sheahan, K.H.; Wahlberg, E.A.; Gilbert, M.P. An overview of GLP-1 agonists and recent cardiovascular outcomes trials. Postgrad. Med. J. 2020, 96, 156–161.
- Ribeiro-Parenti, L.; Jarry, A.-C.; Cavin, J.-B.; Willemetz, A.; Le Beyec, J.; Sannier, A.; Benadda, S.; Pelletier, A.-L.; Hourseau, M.; Léger, T.; et al. Bariatric surgery induces a new gastric mucosa phenotype with increased functional glucagon-like peptide-1 expressing cells. Nat. Commun. 2021, 12, 110.
- Piché, M.E.; Tchernof, A.; Després, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500.
- Sharma, D.; Verma, S.; Vaidya, S.; Kalia, K.; Tiwari, V. Recent updates on GLP-1 agonists: Current advancements & challenges. Biomed. Pharmacother. 2018, 108, 952–962.
- Blair, M. Diabetes Mellitus Review. Urol. Nurs. 2016, 36, 27–36.
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2017, 66, 241–255.
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742.
- Grill, H.J. A Role for GLP-1 in Treating Hyperphagia and Obesity. Endocrinology 2020, 161.
- D’Alessio, D. Is GLP-1 a hormone: Whether and When? J. Diabetes Investig. 2016, 7 (Suppl. 1), 50–55.
- Kanoski, S.E.; Hayes, M.R.; Skibicka, K.P. GLP-1 and weight loss: Unraveling the diverse neural circuitry. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R885–R895.
- Shi, Q.; Wang, Y.; Hao, Q.; Vandvik, P.O.; Guyatt, G.; Li, J.; Chen, Z.; Xu, S.; Shen, Y.; Ge, L.; et al. Pharmacotherapy for adults with overweight and obesity: A systematic review and network meta-analysis of randomised controlled trials. Lancet 2021.
- Athauda, D.; Foltynie, T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: Mechanisms of action. Drug Discov. Today 2016, 21, 802–818.
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807.
- Puschmann, A. New Genes Causing Hereditary Parkinson’s Disease or Parkinsonism. Curr. Neurol. Neurosci. Rep. 2017, 17, 66.
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675.
- Athauda, D.; Maclagan, K.; Budnik, N.; Zampedri, L.; Hibbert, S.; Skene, S.S.; Chowdhury, K.; Aviles-Olmos, I.; Limousin, P.; Foltynie, T. What Effects Might Exenatide have on Non-Motor Symptoms in Parkinson’s Disease: A Post Hoc Analysis. J. Parkinsons Dis. 2018, 8, 247–258.
- Kim, D.S.; Choi, H.I.; Wang, Y.; Luo, Y.; Hoffer, B.J.; Greig, N.H. A New Treatment Strategy for Parkinson’s Disease through the Gut-Brain Axis: The Glucagon-Like Peptide-1 Receptor Pathway. Cell Transplant. 2017, 26, 1560–1571.
- Glotfelty, E.J.; Olson, L.; Karlsson, T.E.; Li, Y.; Greig, N.H. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert Opin. Investig. Drugs 2020, 29, 595–602.
- Grieco, M.; Giorgi, A.; Gentile, M.C.; d’Erme, M.; Morano, S.; Maras, B.; Filardi, T. Glucagon-Like Peptide-1: A Focus on Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1112.
- Hölscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348.
- Mulvaney, C.A.; Duarte, G.S.; Handley, J.; Evans, D.J.; Menon, S.; Wyse, R.; Emsley, H.C. GLP-1 receptor agonists for Parkinson’s disease. Cochrane Database Syst. Rev. 2020, 7, CD012990.
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145–146, 98–120.
- Bifari, F.; Manfrini, R.; Dei Cas, M.; Berra, C.; Siano, M.; Zuin, M.; Paroni, R.; Folli, F. Multiple target tissue effects of GLP-1 analogues on non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Pharmacol. Res. 2018, 137, 219–229.
- Brunner, K.T.; Henneberg, C.J.; Wilechansky, R.M.; Long, M.T. Nonalcoholic Fatty Liver Disease and Obesity Treatment. Curr. Obes. Rep. 2019, 8, 220–228.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20.
- Oseini, A.M.; Sanyal, A.J. Therapies in non-alcoholic steatohepatitis (NASH). Liver. Int. 2017, 37 (Suppl. 1), 97–103.
- Samson, S.L.; Garber, A.J. Metabolic syndrome. Endocrinol. Metab. Clin. N. Am. 2014, 43, 1–23.
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid. Med. Cell Longev. 2019, 2019, 8267234.
- Batista, A.F.; Bodart-Santos, V.; De Felice, F.G.; Ferreira, S.T. Neuroprotective Actions of Glucagon-Like Peptide-1 (GLP-1) Analogues in Alzheimer’s and Parkinson’s Diseases. CNS Drugs 2019, 33, 209–223.
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70.
- Serý, O.; Povová, J.; Míšek, I.; Pešák, L.; Janout, V. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: A review. Folia Neuropathol. 2013, 51, 1–9.
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255.
- Yildirim Simsir, I.; Soyaltin, U.E.; Cetinkalp, S. Glucagon like peptide-1 (GLP-1) likes Alzheimer’s disease. Diabetes Metab. Syndr. 2018, 12, 469–475.
- Boccardi, V.; Murasecco, I.; Mecocci, P. Diabetes drugs in the fight against Alzheimer’s disease. Ageing Res. Rev. 2019, 54, 100936.
- Boyle, J.G.; Livingstone, R.; Petrie, J.R. Cardiovascular benefits of GLP-1 agonists in type 2 diabetes: A comparative review. Clin. Sci. (Lond.) 2018, 132, 1699–1709.
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766.
- Kaltenboeck, A.; Harmer, C. The neuroscience of depressive disorders: A brief review of the past and some considerations about the future. Brain Neurosci. Adv. 2018, 2, 2398212818799269.
- Kim, Y.K.; Kim, O.Y.; Song, J. Alleviation of Depression by Glucagon-Like Peptide 1 Through the Regulation of Neuroinflammation, Neurotransmitters, Neurogenesis, and Synaptic Function. Front. Pharmacol. 2020, 11, 1270.
- Lima-Ojeda, J.M.; Rupprecht, R.; Baghai, T.C. Neurobiology of depression: A neurodevelopmental approach. World J. Biol. Psychiatry 2018, 19, 349–359.
- Stanners, M.N.; Barton, C.A.; Shakib, S.; Winefield, H.R. Depression diagnosis and treatment amongst multimorbid patients: A thematic analysis. BMC Fam. Pract. 2014, 15, 124.
- Weina, H.; Yuhu, N.; Christian, H.; Birong, L.; Feiyu, S.; Le, W. Liraglutide attenuates the depressive- and anxiety-like behaviour in the corticosterone induced depression model via improving hippocampal neural plasticity. Brain Res. 2018, 1694, 55–62.
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 277–284.